PHLOWER: Inferring Complex Cell Differentiation Trajectories from Multimodal Single-Cell Data
The PHLOWER method represents a significant advance in computational trajectory inference, leveraging the harmonic component of the Hodge decomposition on simplicial complexes to reconstruct complex, multi-branching cell differentiation trees from...
PHLOWER: Inferring Complex Cell Differentiation Trajectories from Multimodal Single-Cell Data
Abstract
The PHLOWER method represents a significant advance in computational trajectory inference, leveraging the harmonic component of the Hodge decomposition on simplicial complexes to reconstruct complex, multi-branching cell differentiation trees from single-cell multimodal data. This article provides a comprehensive examination of PHLOWER, from its mathematical foundations in the Hodge Laplacian to its practical applications in identifying transcriptional regulators and characterizing disease processes. Through benchmarking against established methods, we demonstrate PHLOWER's superior performance in recovering complex tree topologies and accurately placing cells within differentiation pathways. For researchers and drug development professionals, this review offers essential insights into implementing, optimizing, and validating PHLOWER across diverse biological contexts.
The Computational Challenge of Cell Differentiation: Why New Methods Are Needed
Cellular differentiation, the process by which unspecialized cells acquire specialized functions, is a cornerstone of embryonic development, tissue homeostasis, and disease pathogenesis [1]. Dissecting the regulatory networks that govern cell fate decisions is crucial for advancing regenerative medicine and therapeutic development. The emergence of single-cell multimodal sequencing technologies has provided unprecedented resolution for studying these processes; however, the computational inference of complex differentiation trajectories from such data presents significant challenges [1] [2]. This application note details how PHLOWER (decomposition of the Hodge Laplacian for inferring trajectories from flows of cell differentiation), a novel computational method leveraging topological data analysis, enables robust inference of complex, multi-branching cell differentiation trees from multimodal single-cell data [1] [3]. We present validated protocols for applying PHLOWER to kidney organoid and other relevant datasets, along with benchmarking data demonstrating its superior performance in reconstructing intricate differentiation pathways and identifying key transcriptional regulators.
The Biological and Technical Challenge
Cellular differentiation is orchestrated by transcription factors (TFs) that bind regulatory DNA regions to control gene expression programs [1]. While multimodal single-cell sequencing can simultaneously measure full expression programs and genome-wide open chromatin in individual cells, experimental protocols dissociate cells, making direct observation of temporal differentiation processes impossible [1]. Computational trajectory inference has therefore become an indispensable tool for reconstructing these dynamics from snapshot data.
The PHLOWER Solution
PHLOWER addresses fundamental limitations in existing trajectory inference methods, which have primarily been validated on simple trees with only 3-9 branches and struggle with complex, multi-branching trajectories [1]. By leveraging the harmonic component of the Hodge decomposition on simplicial complexes (SCs) - higher-order generalizations of graphs that incorporate nodes, edges, triangles, and other structures - PHLOWER directly embeds cell differentiation events and pathways to enable precise detection of complex branching topologies [1] [3].
PHLOWER Methodology and Protocol
Theoretical Foundation
PHLOWER utilizes the discrete Hodge Laplacian (HL) on simplicial complexes, which generalizes the graph Laplacian used in methods like diffusion maps [1]. The spectral decomposition of the HL decomposes edge flows into gradient-free, curl-free, and harmonic components [1] [3]. PHLOWER specifically exploits the harmonic eigenvectors associated with topological "holes" in the simplicial complex, which correspond to cell differentiation tree branches in gene expression space [1].
Step-by-Step Computational Protocol
Protocol 1: Inferring Differentiation Trees with PHLOWER
Objective: Reconstruct complex, multi-branching cell differentiation trajectories from single-cell multimodal data.
Input Requirements:
- Single-cell multimodal data (e.g., scRNA-seq + scATAC-seq) as count matrices
- Cell annotations (optional)
- Computational environment: Standard desktop (16-40 GB RAM recommended)
Procedure:
Data Preprocessing and Graph Construction
- Format input data into an m × n matrix (m observations/cells × n features/genes) [2].
- Construct a graph representation where nodes represent individual cells and edges represent potential differentiation events based on transcriptional similarity.
Pseudotime Estimation and Terminal Cell Identification
- Calculate the graph Laplacian to estimate pseudotime values for all cells.
- Identify progenitor cells (low pseudotime) and terminal differentiated cells (high pseudotime) using established algorithms [1].
Simplicial Complex Formation
- Perform Delaunay triangulation to transform the directed branching process into a simplicial complex.
- Connect terminal differentiated cells with progenitor cells to create topological "holes" representing main differentiation trajectories.
Hodge Decomposition and Harmonic Analysis
- Perform Hodge decomposition of the edge-flow space on the simplicial complex.
- Extract harmonic components to generate:
- Edge-level embeddings (representing individual cell differentiation events)
- Trajectory embeddings (representing complete cell differentiation paths)
Tree Reconstruction and Validation
- Utilize harmonic embeddings to delineate major differentiation trajectories.
- Reconstruct complex cell differentiation trees from trajectory embeddings.
- Validate topology using biological knowledge and benchmark metrics.
Troubleshooting Notes:
- For large datasets (>10,000 cells), apply downsampling to 30% of cells to reduce processing time by ~8.6x and memory usage by approximately one-sixth while maintaining topological accuracy [1].
- Computational requirements typically range from 0.5-12 hours and 12-40 GB memory for datasets of 3,700-18,000 cells on a standard desktop [1].
Experimental Validation Protocol
Protocol 2: Validation of Inferred Trajectories in Kidney Organoids
Objective: Experimentally validate PHLOWER-predicted differentiation trajectories and identify transcription factors regulating off-target cells in kidney organoids [1] [3].
Research Reagent Solutions:
| Reagent/Category | Specific Example | Function in Protocol |
|---|---|---|
| Single-Cell Multimodal Kit | 10x Genomics Multiome ATAC + Gene Expression | Simultaneous measurement of gene expression and open chromatin in individual cells [1] |
| Organoid Culture Media | Kidney organoid differentiation media | Support development and maintenance of kidney organoids containing target cell types [3] |
| Cell Staining Reagents | Immunofluorescence antibodies for kidney markers (e.g., WT1, LTL) | Identification and validation of specific kidney cell types and off-target cells [3] |
| TF Inhibition/Activation | Small molecule inhibitors/activators of predicted TFs | Functional validation of transcription factors identified as key regulators [1] |
| Bioinformatics Tools | R/Python packages for single-cell analysis (Seurat, Scanpy) | Data preprocessing, integration, and independent validation of results [1] |
Procedure:
- Generate kidney organoids using established differentiation protocols.
- Harvest organoids at multiple timepoints for single-cell multimodal sequencing.
- Apply PHLOWER to infer differentiation trajectories and identify branching points.
- Validate predicted cell states spatially using immunofluorescence for key markers.
- Perturb predicted regulatory TFs using pharmacological inhibitors/activators or genetic approaches.
- Re-sequence perturbed organoids to confirm trajectory alterations.
Expected Outcomes: Identification of transcription factors regulating off-target cell populations in kidney organoids, enabling protocol optimization for improved purity [1] [3].
Performance Benchmarking and Applications
Quantitative Performance Assessment
Table 1: Benchmarking PHLOWER Against Competing Methods on Simulated Data (DLA Trees with 5-18 Branches) [1]
| Method | Tree Topology Recovery (HIM) | Cell Location within Branch (Correlation) | Cell Allocation to Branches (F1) | Overall Accuracy |
|---|---|---|---|---|
| PHLOWER | Best performance | Best performance | Best performance | Best performance |
| PAGA | Second best | Moderate | Second best | Second best |
| RaceID | Third best | Third best | Third best | Third best |
| Monocle3 | Moderate | Moderate | Moderate | Moderate |
| Slingshot | Lower performance | Second best | Lower performance | Lower performance |
Table 2: Benchmarking on Real scRNA-seq Data (33 Dynverse Datasets) [1]
| Method | Tree Structure Similarity (HIM) | Cell Location within Branch (Correlation) | Cell Allocation to Branches (F1) | Overall Accuracy |
|---|---|---|---|---|
| PHLOWER | Best performance | Best performance | Best performance | Best performance |
| Monocle3 | Second best | Second best | Moderate | Second best |
| pCreode | Moderate | Moderate | Moderate | Third best |
| Slingshot | Lower performance | Third best | Second best | Moderate |
Application to Disease Modeling and Drug Development
PHLOWER enables precise characterization of disease-related differentiation processes, such as aberrant cell fate decisions in cancer or defective differentiation in genetic disorders [1]. By identifying key transcriptional regulators of these pathological transitions, researchers can prioritize therapeutic targets for small molecule screening or cellular reprogramming approaches. The method's ability to leverage multimodal data is particularly valuable for understanding the interplay between chromatin accessibility and gene expression changes in disease states.
Visualizing the PHLOWER Workflow
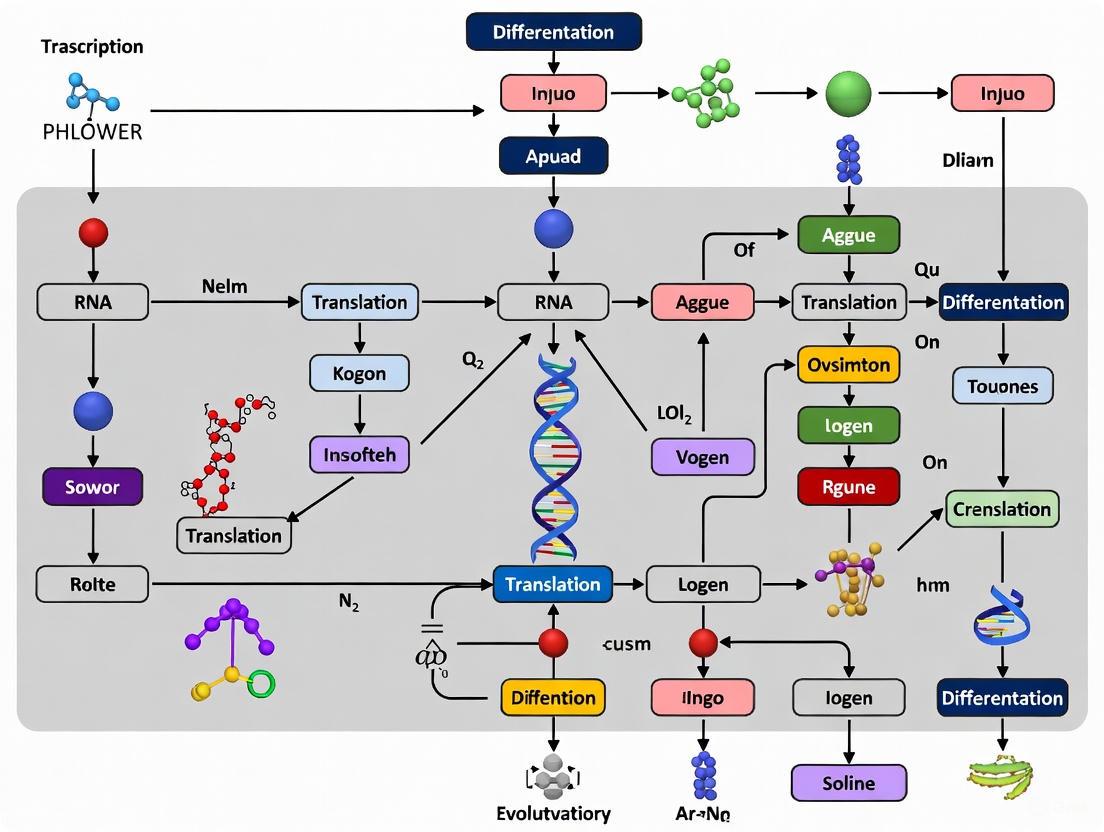
Figure 1: PHLOWER computational workflow for inferring cell differentiation trajectories from single-cell multimodal data.
Figure 2: PHLOWER identifies complex branching trajectories and off-target cells in differentiation processes, enabling improved protocol optimization.
Limitations of Current Trajectory Inference Methods with Complex Tree Structures
Trajectory inference (TI) is a foundational computational technique in single-cell omics analysis that reconstructs dynamic cellular progression, such as differentiation, from static snapshot data by ordering cells along a pseudotime axis [4]. The field has evolved from methods handling simple linear paths to those addressing branching topologies, yet a significant challenge remains in accurately inferring complex, multi-branching trees [1] [4]. This application note delineates the specific limitations of current TI methodologies when confronted with such intricate structures, framing the discussion within the broader research context of the recently developed PHLOWER method, which leverages the Hodge Laplacian and simplicial complexes to address these challenges [1].
Key Limitations of Current TI Methods
The performance gap of existing TI methods on complex trees manifests in several key areas, from structural inference to computational feasibility.
Inaccurate Topology Reconstruction
Many established TI methods exhibit a tendency to underestimate the size and complexity of differentiation trees. Benchmarking studies reveal that while some methods perform adequately on simpler bifurcating trajectories, they struggle with multifurcations and trees containing higher numbers of branches (e.g., 5 to 18 branches) [1]. For instance, in analyses of both simulated and real single-cell RNA-sequencing (scRNA-seq) data, methods like Slingshot were observed to infer oversimplified tree structures, whereas only PHLOWER, PAGA, and Monocle3 were capable of recovering more complex topologies [1]. This often results in the obscuration of elusive lineages and less populous cell fates, which can be biologically critical yet computationally elusive [5].
Table 1: Performance of TI Methods on Complex Tree Inference
| Method | Performance on Simulated Complex Trees | Performance on Real scRNA-seq Trees | Key Limitation |
|---|---|---|---|
| PHLOWER | Top performer in topology recovery & cell allocation [1] | Top performer in structure similarity & branch allocation [1] | High computational demand for large datasets [1] |
| PAGA | Runner-up in topology recovery [1] | Runner-up in structure similarity [1] | Performance varies with tree structure type [1] |
| Monocle3 | Runner-up in cell branch allocation [1] | Runner-up in cell location & accuracy [1] | May not capture full complexity in some datasets [1] |
| Slingshot | Lower performance in complex tree benchmarks [1] | Good performance on simpler bifurcations [1] | Tends to underestimate tree size and complexity [1] |
| RaceID | Runner-up in simulated data accuracy [1] | Lower performance on real data [1] | Inconsistent performance between simulated vs. real data [1] |
| VIA | Captures complex, multi-furcating, and cyclic topologies [5] | Generalizes across transcriptomic, epigenomic, and multi-omic data [5] | Not included in the primary benchmark cited [1] |
Scalability and Computational Constraints
The computational burden associated with processing large-scale single-cell data presents another major hurdle. As datasets grow to encompass millions of cells, as in whole-organism atlases, many TI methods require extensive dimensionality reduction or multiple rounds of subsampling, which can compromise global connectivity information and obscure fine-grained trajectories [5]. For example, applying PHLOWER to a neurogenesis dataset of approximately 18,000 cells required 12-40 GB of memory and 0.5-12 hours on a standard desktop [1]. While a downsampling variant can mitigate this, it underscores the inherent computational intensity of high-resolution trajectory inference on complex trees.
Challenges with Multimodal and Multi-Condition Data
Modern single-cell technologies increasingly provide multimodal measurements, simultaneously capturing transcriptomic, epigenomic, and proteomic data from the same cell. There is a noted lack of computational approaches capable of inferring complex branching trajectories from such integrated data [1]. Furthermore, analyzing trajectories across multiple conditions (e.g., healthy vs. diseased, wild-type vs. knock-out) introduces additional challenges. Methods must determine whether to fit a common trajectory or separate ones for each condition (a problem of differential topology), and how to test for differences in progression or fate selection along lineages [6]. Most TI methods lack this functionality natively, creating an analytical gap.
Experimental Benchmarking and Evaluation Protocols
Protocol for Benchmarking TI Methods on Complex Trees
Objective: To quantitatively evaluate and compare the performance of various trajectory inference methods on datasets with known, complex tree-like topologies.
Materials:
- Simulated Data: Utilize the diffusion-limited aggregation (DLA) model to generate complex differentiation tree datasets with a high number of branches (e.g., 5 to 18 total branches) [1].
- Real scRNA-seq Data: Select single-rooted tree-structured datasets from curated resources like Dynverse that contain at least three branches [1].
- Software: TI methods to be evaluated (e.g., PHLOWER, PAGA, Monocle3, Slingshot, TSCAN, VIA).
Procedure:
- Data Preparation: Preprocess all datasets (simulated and real) using a standardized pipeline, including normalization and quality control.
- Trajectory Inference: Apply each TI method to the datasets according to their standard workflows. For methods requiring a root cell, specify the same progenitor population across all methods for a fair comparison.
- Performance Evaluation: Calculate the following metrics using a framework like Dynverse [1]:
- Hamming–Ipsen–Mikhailov (HIM): Measures the similarity between the inferred and ground-truth tree structures.
- Correlation: Assesses the correctness of cell ordering within each branch.
- F1 Branches: Evaluates the accuracy of assigning cells to the correct branches.
- F1 Milestones: Evaluates the accuracy of assigning cells to the correct branching points.
- Aggregated Accuracy: The average of the four above metrics.
- Computational Profiling: Record the runtime and peak memory usage for each method on each dataset.
Analysis:
- Rank methods based on their aggregated accuracy and individual metric scores.
- Stratify performance based on tree type (bifurcation, multifurcation, complex trees) to identify method-specific strengths.
- Correlate computational requirements with dataset size and complexity.
Protocol for Assessing Differential Topology Across Conditions
Objective: To determine if an underlying developmental trajectory differs fundamentally between two or more biological conditions (e.g., control vs. perturbation).
Materials:
- scRNA-seq Data: Data from multiple conditions (e.g., wild-type and knock-out).
- Software: A multi-condition analysis tool such as
condiments[6].
Procedure:
- Data Integration: Integrate datasets from all conditions into a shared low-dimensional space.
- Trajectory Inference: Input the integrated data into a TI method (e.g., Slingshot) to infer an initial consensus trajectory.
- Topology Test: Using the
condimentsworkflow, perform a statistical test for differential topology. This test assesses whether the trajectory structure itself is significantly different between conditions [6]. - Interpretation:
- If the test indicates significant differential topology, infer separate trajectories for each condition.
- If not, a common trajectory can be used for subsequent analyses of differential progression and fate selection.
PHLOWER (decomposition of the Hodge Laplacian for inferring trajectories from flows of cell differentiation) represents a novel approach designed to address the limitation of inferring complex trees [1]. Its workflow and key differentiators are summarized below.
Diagram 1: The PHLOWER analytical workflow for inferring complex trajectories from single-cell data.
Research Reagent Solutions
Table 2: Key Computational Tools and Data for Trajectory Inference Research
| Resource Name | Type | Primary Function in TI Research |
|---|---|---|
| PHLOWER | Software Package | Infers complex, multi-branching differentiation trees from single-cell multimodal data using Hodge Laplacian decomposition [1]. |
| VIA | Software Package | Generalizable TI for complex topologies (trees, cycles, disconnected) on large-scale and multi-omic data [5]. |
| condiments | Software Package / R Package | Provides a framework for inference and interpretation of trajectories across multiple conditions, testing for differential topology, progression, and fate selection [6]. |
| Dynverse | Software Suite / Benchmarking Platform | A curated platform and dataset repository for benchmarking and evaluating trajectory inference methods [1]. |
| DLA Simulated Trees | Synthetic Data | Generates complex benchmark differentiation tree datasets with a controllable number of branches for method validation [1]. |
| tradeSeq | Software Package / R Package | Fits gene expression smoothers along trajectories and identifies differentially expressed genes across lineages [6]. |
Cellular differentiation is a fundamental process in development and disease, yet tracking how an individual cell changes over time experimentally is impossible because single-cell sequencing protocols dissociate cells from their tissue context [1]. Computational trajectory inference has therefore become a critical tool for reconstructing these differentiation pathways from snapshots of single-cell data [1]. While multimodal single-cell sequencing can measure both gene expression and genome-wide open chromatin—providing a unique resource to understand the interplay between chromatin and gene expression during differentiation—existing computational approaches have struggled with complex, multi-branching trees [1]. Prior to PHLOWER, methods were primarily evaluated on small trees with only four to five differentiation branches, leaving a gap in capabilities for analyzing more complex biological systems [1].
PHLOWER (decomposition of the Hodge Laplacian for inferring trajectories from flows of cell differentiation) addresses these limitations by introducing a novel mathematical framework based on simplicial complexes and the Hodge Laplacian [1]. This approach enables researchers to infer complex differentiation trajectories from multimodal single-cell data, facilitating the identification of key transcriptional regulators and providing insights into developmental biology and disease mechanisms [1].
Mathematical Framework and Methodological Principles
Core Theoretical Foundations
PHLOWER is built upon several advanced mathematical concepts that generalize traditional graph-based approaches:
Simplicial Complexes (SCs): PHLOWER represents single-cell multimodal data using simplicial complexes, which are higher-order generalizations of graphs that allow not only for nodes (0-simplices) and edges (1-simplices) but also triangles (2-simplices) and other higher-order structures [1]. This representation captures more complex topological relationships between cells than standard graph methods.
Hodge Laplacian (HL): The discrete Hodge Laplacian on simplicial complexes represents a generalization of the well-known graph Laplacian used in methods like diffusion maps [1]. While the graph Laplacian is a zero-order Hodge Laplacian, the full HL operates on higher-order structures in the simplicial complex.
Hodge Decomposition: The spectral decomposition of the Hodge Laplacian decomposes edge flows into three orthogonal components: gradient-free, curl-free, and harmonic components [1]. PHLOWER specifically leverages the harmonic eigenvectors of the Hodge Laplacian, which are associated with holes in the simplicial complex that correspond to cell differentiation tree branches in the gene expression space [1].
Algorithmic Workflow
The PHLOWER algorithm transforms single-cell data into differentiation trajectories through these key steps:
Graph Representation: Single-cell data is first represented as a graph where nodes represent individual cells and edges represent potential differentiation events [1].
Pseudotime Estimation: Using the graph Laplacian, PHLOWER estimates pseudotime of cells and identifies progenitor cells (low pseudotime) and terminal differentiated cells (high pseudotime), similar to existing trajectory inference methods [1].
Simplicial Complex Construction: A Delaunay triangulation connects terminal differentiated cells with progenitor cells. This connection creates a "hole" for every main trajectory in the graph, which becomes mathematically detectable through the Hodge decomposition [1].
Hodge Decomposition: The algorithm performs a Hodge decomposition of the edge-flow space on the simplicial complex. The harmonic components of this decomposition provide both edge-level embeddings (where each point represents a cell differentiation event) and trajectory embeddings (where each point represents a cell differentiation trajectory) [1].
Tree Reconstruction: These natural representations of cell differentiation facilitate the estimation of the underlying differentiation trees, enabling the identification of complex branching patterns [1].
The following diagram illustrates the core computational workflow of PHLOWER:
Benchmarking Performance and Comparative Analysis
Evaluation Framework and Metrics
PHLOWER was rigorously evaluated against leading trajectory inference methods using both simulated datasets and real single-cell RNA-sequencing (scRNA-seq) data [1]. The benchmarking utilized:
- Simulated Data: Ten complex differentiation tree datasets generated using diffusion-limited aggregation (DLA) trees with 5 to 18 total branches [1].
- Real scRNA-seq Data: 33 datasets from the Dynverse benchmarking resource containing single-rooted tree structures with at least three branches [1].
- Competing Methods: PAGA tree, Monocle3, cellTree, pCreode, Slice, RaceID, Slingshot, TSCAN, MST, Elpigraph, and STREAM—representing the top approaches from previous evaluations [1].
Performance was assessed using multiple metrics from the Dynverse framework: Hamming–Ipsen–Mikhailov (HIM) for tree structure similarity, correlation for location of cells within branches, F1 branches for cell allocation to branches, F1 milestones for cell allocation to branching points, and an overall accuracy calculated as the average of these four metrics [1].
Quantitative Performance Results
Table 1: Benchmarking Results on Simulated Data with Complex Branching Trees (5-18 branches)
| Method | Tree Structure (HIM) | Cell Location (Correlation) | Cell to Branch (F1) | Overall Accuracy |
|---|---|---|---|---|
| PHLOWER | Best Performance | Best Performance | Best Performance | Best Performance |
| PAGA | Second Best | Moderate | High | High |
| RaceID | High | High | High | High |
| Monocle3 | Moderate | Moderate | High | Moderate |
| TSCAN | Moderate | High | Moderate | Moderate |
Table 2: Benchmarking Results on Real scRNA-seq Data
| Method | Tree Structure (HIM) | Cell Location (Correlation) | Cell to Branch (F1) | Overall Accuracy |
|---|---|---|---|---|
| PHLOWER | Best Performance | Best Performance | Best Performance | Best Performance |
| Monocle3 | High | High | High | High |
| pCreode | Moderate | Moderate | Moderate | High |
| Slingshot | Moderate | High | High | Moderate |
| PAGA | High | Moderate | Moderate | Moderate |
Table 3: Performance Stratification by Tree Complexity in Real Data
| Method | Bifurcation Trees | Multifurcation Trees | Complex Trees |
|---|---|---|---|
| PHLOWER | High Performance | High Performance | High Performance |
| Slingshot | High Performance | Moderate | Low Performance |
| Monocle3 | Moderate | High Performance | High Performance |
| PAGA | Moderate | Moderate | High Performance |
The benchmarking revealed that PHLOWER consistently achieved top performance across both simulated and real datasets, demonstrating particular strength in recovering complex tree topologies and accurately allocating cells to their correct positions within differentiation branches [1]. Notably, while some methods like Slingshot performed well on simpler bifurcating trees, they tended to underestimate the complexity of larger trees, whereas PHLOWER, Monocle3, and PAGA maintained better performance on complex structures [1].
Experimental Protocols and Implementation
Computational Requirements and Optimization
Implementation of PHLOWER requires consideration of computational resources, particularly for larger datasets:
- Hardware Requirements: For standard desktop computing, PHLOWER required 0.5–12 hours and 12–40 GB of memory when analyzing pancreas progenitor (~3,700 cells) and neurogenesis (~18,000 cells) datasets [1].
- Scalability Optimization: A time-efficient variant using cell downsampling (30% of cells) achieved 8.6 times faster processing and used one-sixth of the memory while still inferring biologically accurate differentiation trees [1].
Protocol: Applying PHLOWER to Multimodal Single-Cell Data
Objective: Reconstruct complex differentiation trajectories from single-cell multimodal data and identify key transcription factors regulating cell fate decisions.
Materials and Reagents:
Table 4: Essential Research Reagent Solutions for PHLOWER Analysis
| Reagent/Resource | Function/Application | Specifications |
|---|---|---|
| Multimodal Single-cell Data | Primary input for trajectory inference | Must include gene expression and chromatin accessibility data |
| Computational Environment | Hardware/software platform for analysis | Minimum 12-40 GB RAM; R/Python environment |
| Cell Annotations | Preliminary cell type identification | Enables progenitor/terminal cell identification |
| Reference Differentiation Trees | Validation of results | Known pathways for biological context |
Step-by-Step Procedure:
Data Preprocessing
- Load multimodal single-cell data (e.g., simultaneous RNA expression and ATAC-seq data)
- Perform quality control, normalization, and batch correction using standard single-cell analysis pipelines
- Integrate multimodal measurements to create a unified cell-by-feature matrix
Initial Graph Construction
- Create a k-nearest neighbor graph from the integrated single-cell data
- Calculate the graph Laplacian and estimate pseudotime using diffusion components
- Identify putative progenitor cells (low pseudotime) and terminal differentiated cells (high pseudotime)
Simplicial Complex Formation
- Perform Delaunay triangulation on the cell embedding
- Connect terminal cells with progenitor cells to introduce topological holes corresponding to potential differentiation trajectories
- Validate simplicial complex construction to ensure biological relevance
Hodge Laplacian Decomposition
- Construct the discrete Hodge Laplacian operator on the simplicial complex
- Perform spectral decomposition to separate edge flows into gradient, curl, and harmonic components
- Extract the harmonic component representing the non-trivial looping flows around topological holes
Trajectory Embedding and Tree Inference
- Generate edge-flow embeddings from harmonic components where each point represents a potential differentiation event
- Create trajectory embeddings where each point represents a complete differentiation path
- Apply clustering algorithms to identify distinct differentiation branches
- Reconstruct the final branching tree structure representing the differentiation process
Biological Validation and Interpretation
- Map key transcription factors along differentiation branches using matched chromatin accessibility data
- Identify regulatory regions that gain or lose accessibility during differentiation
- Validate identified trajectories using known marker genes and previously established differentiation pathways
The following workflow diagram illustrates the key experimental steps in the PHLOWER protocol:
Application Notes and Biological Validation
Case Study: Kidney Organoid Development
PHLOWER was validated using kidney organoid multimodal and spatial single-cell data, demonstrating its power in both inferring complex trees and identifying transcription factors regulating off-target cells in kidney organoids [1]. The method successfully:
- Reconstructed the complex branching trajectory of nephron development in organoid culture
- Identified key transcription factors associated with specific lineage decisions
- Detected off-target cell populations and predicted their regulatory mechanisms
- Integrated spatial information when available to enhance trajectory accuracy
Protocol: Troubleshooting and Quality Control
Common Issues and Solutions:
- Poor Tree Structure: If the resulting tree lacks clear branching structure, verify the quality of the input data and consider adjusting the parameters for simplicial complex construction.
- Computational Limitations: For very large datasets (>20,000 cells), implement the downsampling approach using 30% of cells to reduce computational requirements while maintaining accuracy [1].
- Biological Plausibility: Always validate inferred trajectories against known biological pathways and marker gene expression patterns.
Quality Control Metrics:
- Compare inferred tree complexity with known biology of the system
- Verify that progenitor cells are correctly positioned at the root of the tree
- Validate that terminal states correspond to known differentiated cell types
- Confirm that pseudotime ordering aligns with expression of maturation markers
Integration with Drug Development and Therapeutic Applications
The ability to accurately reconstruct differentiation trajectories has significant implications for drug development and therapeutic interventions. PHLOWER's capacity to identify transcription factors regulating cell fate decisions provides potential targets for:
- Cellular Reprogramming: Identifying key regulators that could be targeted to redirect cell differentiation for regenerative medicine applications
- Disease Modeling: Understanding how differentiation pathways are altered in disease states, particularly cancer and developmental disorders
- Organoid Optimization: Identifying and correcting off-target cells in organoid cultures to improve disease modeling and drug screening platforms
- Toxicology Studies: Tracing the impact of compounds on differentiation pathways to assess developmental toxicity
PHLOWER represents a significant advancement in computational trajectory inference, particularly for complex, multi-branching differentiation processes. By leveraging the mathematical framework of Hodge decomposition on simplicial complexes, it enables researchers to extract more accurate and detailed differentiation pathways from multimodal single-cell data, with broad applications in basic research, drug discovery, and therapeutic development.
The PHLOWER method represents a significant advancement in computational trajectory analysis for inferring cell differentiation trees from single-cell multimodal data. A primary challenge in the field has been the prediction of complex, multi-branching trees from such data, which previous approaches struggled to address with scalability [7]. PHLOWER addresses this fundamental limitation through its novel application of the harmonic component of the Hodge decomposition on simplicial complexes, moving beyond traditional graph-based representations to create more accurate trajectory embeddings [7] [3]. This mathematical framework enables researchers to directly represent cell differentiation processes, facilitating the detection of complex branching events with high precision that were previously difficult or impossible to identify.
At its core, PHLOWER leverages the discrete Hodge Laplacian (HL), which generalizes the well-known graph Laplacian used in methods like diffusion maps. While the graph Laplacian provides a matrix representation where samples are vertices and distances are edge weights, the Hodge Laplacian operates on higher-order topological structures [7]. This allows PHLOWER to capture more complex relationships in single-cell data that traditional graph-based methods miss, particularly when dealing with multifurcating differentiation trees common in developmental biology and disease processes.
Technical Specifications and Mathematical Foundation
Simplicial Complex Representations in Single-Cell Data
PHLOWER represents single-cell data as a simplicial complex (SC), which generalizes standard graph structures to include higher-order relationships. A simplicial complex consists of:
- Nodes (0-simplices): Represent individual cells in the dataset
- Edges (1-simplices): Represent potential cell differentiation events between cells
- Triangles (2-simplices): Represent higher-order relationships among triplets of cells [7]
This representation differs fundamentally from traditional graph-based approaches because it preserves topological information about "holes" or cycles in the data, which correspond to branching events in differentiation trajectories. The connection between terminal differentiated cells (high pseudotime) and progenitor cells (low pseudotime) creates a topological hole for every main trajectory in the graph, which the Hodge decomposition can subsequently identify [7].
Hodge Decomposition and Harmonic Components
The Hodge decomposition facilitated by PHLOWER operates on the edge-flow space of the simplicial complex, decomposing it into three orthogonal components:
- Gradient component: Representing conservative flows
- Curl component: Representing rotational flows
- Harmonic component: Representing globally non-trivial flows that are neither gradient nor curl [7]
PHLOWER specifically focuses on the harmonic eigenvectors of the Hodge Laplacian, as these are mathematically guaranteed to be associated with holes in the simplicial complex. In the context of single-cell data, these holes directly correspond to branching events in cell differentiation trees [7]. The harmonic components provide two crucial types of embeddings:
- Edge-level embeddings: Where each point represents a cell differentiation event
- Trajectory embeddings: Where each point represents an entire cell differentiation path [7]
Table 1: Key Mathematical Components of the PHLOWER Framework
| Component | Mathematical Description | Biological Interpretation |
|---|---|---|
| Simplicial Complex | Higher-order generalization of graphs (nodes, edges, triangles) | Representation of cellular relationships and potential differentiation paths |
| Hodge Laplacian | Operator on simplicial complexes generalizing graph Laplacian | Captures topological structure of cell differentiation landscape |
| Harmonic Component | Null space of Hodge Laplacian | Identifies branching points and distinct differentiation trajectories |
| Edge-Flow Embedding | Embedding space where points represent differentiation events | Maps potential transitions between cell states |
| Trajectory Embedding | Embedding space where points represent differentiation paths | Reconstructs entire lineages from progenitor to terminal cells |
Benchmarking Performance and Quantitative Validation
Experimental Design for Method Evaluation
PHLOWER underwent rigorous benchmarking against established trajectory inference methods using both simulated datasets and real single-cell RNA-sequencing (scRNA-seq) data. The evaluation framework included:
- Simulated datasets: Ten complex differentiation tree datasets generated using diffusion-limited aggregation (DLA) trees with 5 to 18 total branches [7]
- Real scRNA-seq datasets: 33 datasets from the Dynverse benchmarking resource containing single-rooted tree structures with at least three branches [7]
- Competing methods: Comparison against PAGA tree, Monocle3, cellTree, pCreode, Slice, RaceID, Slingshot, TSCAN, MST, Elpigraph, and STREAM [7]
Performance was evaluated using the Dynverse framework metrics, including tree structure similarity (Hamming-Ipsen-Mikhailov, HIM), correlation of cell locations within branches, F1 score for cell allocation to branches, F1 score for cell allocation to branching points, and overall accuracy as the average of these metrics [7].
Quantitative Performance Results
Table 2: Benchmarking Results of PHLOWER Against Competing Methods
| Performance Metric | Simulated Data (PHLOWER Rank) | Real scRNA-seq Data (PHLOWER Rank) | Top Competing Methods |
|---|---|---|---|
| Tree Topology Recovery | 1st | 1st | PAGA, RaceID, Monocle3 |
| Cell Location within Branches | 1st | 1st | TSCAN, RaceID, Monocle3 |
| Cell Allocation to Branches | 1st | 1st | Monocle3, PAGA |
| Cell Allocation to Branching Points | 1st | 1st | RaceID, Monocle3, PAGA |
| Overall Accuracy | 1st | 1st | PAGA, RaceID, Monocle3 |
The benchmarking results demonstrate PHLOWER's consistent superiority across all evaluation metrics. On simulated data, PHLOWER achieved the highest performance in tree topology recovery, cell location accuracy, and branch allocation, followed by PAGA, RaceID, and Monocle3 as the next best performers [7]. Similarly, for real scRNA-seq data, PHLOWER maintained top performance in structure similarity, cell location, and branch allocation, with Monocle3, pCreode, and Slingshot as runners-up [7].
Notably, the performance analysis revealed that method performance varies based on tree complexity. While some approaches like Slingshot perform relatively better on simpler bifurcating trees, PHLOWER, Monocle3, and PAGA demonstrate superior performance on complex multifurcating structures [7]. This distinction is crucial for researchers studying complex differentiation processes with multiple potential lineage branches.
Experimental Protocols and Implementation
Standard PHLOWER Workflow Protocol
Figure 1: Standard computational workflow for implementing PHLOWER analysis, showing the sequence from data input to tree reconstruction.
Protocol Steps:
- Input Data Preparation: Format single-cell multimodal data (e.g., simultaneous gene expression and chromatin accessibility measurements) into a standardized matrix format [7].
- Graph Construction: Create a k-nearest neighbor graph representation of the single-cell data where nodes represent cells and edge weights represent cellular similarities [7].
- Pseudotime Estimation: Calculate pseudotime values for all cells using the graph Laplacian, similar to approaches used in diffusion maps and other trajectory inference methods [7].
- Progenitor and Terminal Cell Identification: Identify putative progenitor cells (low pseudotime) and terminally differentiated cells (high pseudotime) based on pseudotime ordering [7].
- Delaunay Triangulation: Perform spatial triangulation to connect cells in the embedding space, creating higher-order relationships beyond simple pairwise connections [7].
- Simplicial Complex Construction: Build the full simplicial complex representation incorporating nodes (cells), edges (differentiation events), and triangles (higher-order relationships) [7].
- Hodge Laplacian Decomposition: Compute the spectral decomposition of the Hodge Laplacian operator on the constructed simplicial complex [7].
- Harmonic Component Extraction: Isolate the harmonic components from the Hodge decomposition, which capture the non-trivial cyclic structures in the data corresponding to branching events [7].
- Trajectory Embedding Generation: Project the harmonic components into lower-dimensional embedding spaces for both edge-flows (differentiation events) and trajectories (differentiation paths) [7].
- Tree Reconstruction: Apply tree-building algorithms to the trajectory embeddings to reconstruct the final cell differentiation tree with complex branching structures [7].
Computational Optimization Protocol
Figure 2: Optimization protocol for handling large-scale single-cell datasets while maintaining analytical performance.
Optimization Protocol: For large datasets (e.g., >10,000 cells), computational requirements can become substantial. The standard PHLOWER implementation required 0.5-12 hours and 12-40 GB of memory for datasets ranging from approximately 3,700 to 18,000 cells using standard desktop computing resources [7]. To address this:
- Cell Downsampling: Implement strategic downsampling to approximately 30% of cells while preserving population structure and rare cell types [7].
- Subsampled Analysis: Apply the standard PHLOWER workflow to the downsampled data.
- Performance Validation: Verify that the inferred tree maintains biological fidelity compared to full-dataset analysis.
This optimized approach demonstrated 8.6 times faster processing and used one-sixth of the memory compared to full analysis while still inferring biologically accurate differentiation trees [7].
Research Reagent Solutions and Computational Tools
Table 3: Essential Computational Tools and Resources for PHLOWER Implementation
| Tool Category | Specific Solutions | Function in PHLOWER Workflow |
|---|---|---|
| Single-cell Multimodal Platforms | 10x Genomics Multiome | Simultaneous measurement of gene expression and chromatin accessibility from single cells [7] |
| Data Processing Tools | Seurat, Scanpy | Preprocessing, quality control, and initial dimensionality reduction of single-cell multimodal data [7] |
| Trajectory Inference Benchmarks | Dynverse | Standardized framework for evaluating trajectory inference method performance [7] |
| Computational Environments | R, Python | Implementation of Hodge Laplacian decomposition and harmonic component analysis [7] |
| Visualization Tools | ggplot2, Matplotlib | Visualization of trajectory embeddings and differentiation tree structures [7] |
Application to Kidney Organoid and Spatial Data
The PHLOWER method has been successfully applied to analyze kidney organoid multimodal and spatial single-cell data, demonstrating its practical utility in complex biological systems. In these applications, PHLOWER not only inferred complex branching trajectories but also identified transcription factors regulating off-target cells in kidney organoids [7] [3]. This capability is particularly valuable for optimizing organoid differentiation protocols and understanding disease-related differentiation processes for potential therapeutic interventions [7].
The application to spatial single-cell data further highlights PHLOWER's versatility, as the method can incorporate spatial neighborhood information into the simplicial complex representation, potentially revealing spatial patterns of differentiation that would be missed in dissociated single-cell data alone [7].
The Growing Importance of Multimodal Single-Cell Data in Differentiation Studies
The emergence of sophisticated single-cell technologies has revolutionized our ability to study cellular differentiation, the fundamental process by which cells transition from multipotent states to specialized fates. While single-cell RNA sequencing (scRNA-seq) has provided unprecedented insights into cellular heterogeneity, it captures only one dimension of the complex regulatory machinery. Multimodal single-cell data represents a significant technological advancement, enabling the simultaneous measurement of multiple molecular modalities—such as transcriptome, chromatin accessibility, and protein expression—from the same cell. This integrated approach provides a more comprehensive view of cell state and identity, capturing the synergistic relationship between gene expression and epigenetic regulation during cell fate decisions.
The analysis of such rich, multi-layered data presents substantial computational challenges, particularly in reconstructing the complex, multi-branching trajectories that characterize differentiation in development, disease, and organoid models. Computational trajectory inference methods must now integrate disparate data types to map the dynamic transitions between cell states with higher fidelity. In this context, the PHLOWER method (decomposition of the Hodge Laplacian for inferring trajectories from flows of cell differentiation) represents a significant innovation. By leveraging the harmonic component of the Hodge decomposition on simplicial complexes, PHLOWER infers trajectory embeddings specifically from single-cell multimodal data, enabling more accurate estimation of underlying differentiation trees and identification of key transcriptional regulators [3] [8].
Core Computational Framework
PHLOWER addresses a critical open challenge in computational biology: predicting complex, multi-branching differentiation trees from multimodal single-cell data. Traditional trajectory inference methods often struggle with the complexity of organogenesis and tissue development, where differentiation pathways branch repeatedly to generate diverse cell types. PHLOWER's mathematical foundation in Hodge decomposition provides a natural framework for representing these complex branching processes by analyzing flows of cell differentiation within simplicial complexes—mathematical structures that generalize graphs to higher dimensions [3].
The method specifically leverages the harmonic component of this decomposition, which captures the global structure of the differentiation flow independent of local noise or artifacts. This approach enables PHLOWER to infer trajectory embeddings that faithfully represent the underlying biological processes, facilitating the construction of accurate differentiation trees from integrated multimodal data. The algorithm's ability to handle multiple data modalities simultaneously allows it to detect coordinated changes in different molecular layers, providing stronger evidence for branching points and transition states than would be possible with unimodal data alone [8].
Key Advantages and Applications
PHLOWER has demonstrated particular utility in evaluating complex biological systems such as kidney organoids, where it successfully inferred multi-branching differentiation trees from multimodal and spatial single-cell data [3]. Beyond trajectory reconstruction, the method enables identification of transcription factors regulating off-target cells in kidney organoids—a crucial application for improving organoid quality and fidelity. This capability to not only map differentiation pathways but also identify their molecular regulators represents a significant advance for both developmental biology and therapeutic applications.
Table 1: Key Features and Applications of the PHLOWER Method
| Feature | Description | Application |
|---|---|---|
| Multimodal Data Integration | Leverages simultaneous measurements of transcriptome, chromatin state, and other modalities | Provides more robust identification of cell states and transitions |
| Complex Tree Inference | Infers multi-branching differentiation trajectories using Hodge decomposition | Maps complex differentiation processes in development and organoid models |
| Regulatory Network Analysis | Identifies transcription factors driving differentiation decisions | Discovers molecular regulators of cell fate, including off-target cells in organoids |
| Spatial Data Compatibility | Incorporates spatial single-cell data when available | Enhances trajectory inference with tissue context information |
Experimental Protocols for Multimodal Trajectory Analysis
Protocol 1: Sample Preparation and Multimodal Data Generation
Objective: Generate high-quality multimodal single-cell data suitable for trajectory inference using PHLOWER.
Materials and Reagents:
- Fresh tissue or organoid samples
- Single-cell dissociation kit (enzyme-based)
- Single-cell multimodal profiling platform (e.g., 10x Multiome, CITE-seq)
- Viability dye
- Phosphate-buffered saline
- BSA
- Nuclei isolation buffer
Procedure:
- Sample Preparation: Dissociate tissue or organoids into single-cell suspensions using optimized enzymatic digestion. Filter through appropriate mesh to remove aggregates.
- Viability Assessment: Stain cells with viability dye and quantify viability using hemocytometer or automated cell counter. Proceed only if viability exceeds 80%.
- Multimodal Library Preparation: Process cells according to chosen multimodal platform protocol. For joint ATAC + RNA profiling:
- Perform tagmentation on isolated nuclei
- Barcode RNA and DNA separately
- Generate cDNA and library for RNA-seq
- Amplify and index ATAC library
- Quality Control: Assess library quality using Bioanalyzer or TapeStation before sequencing.
- Sequencing: Sequence libraries following platform-specific recommendations for read depth and distribution.
Troubleshooting Tips:
- If cell viability is low, optimize dissociation conditions and reduce processing time
- If library complexity is low, increase cell input and verify reagent freshness
- If RNA quality is poor, use RNA stabilizers during dissociation
Protocol 2: Computational Analysis with PHLOWER
Objective: Process multimodal single-cell data to infer differentiation trajectories using PHLOWER.
Input Data Requirements:
- Gene expression matrix
- Chromatin accessibility matrix
- Cell metadata
Software and Dependencies:
- PHLOWER implementation
- Python with scientific computing stack
- Single-cell analysis tools
Procedure:
- Data Preprocessing:
- Filter cells based on quality metrics
- Normalize gene expression matrix
- Process chromatin accessibility peaks
- Integrate modalities using dimensionality reduction
PHLOWER Analysis:
- Construct simplicial complex representation
- Compute Hodge Laplacian
- Perform harmonic decomposition
- Infer trajectory embeddings
Trajectory Visualization:
- Generate force-directed layout
- Plot differentiation tree
- Annotate branches with cell type labels
Regulator Identification:
- Correlate transcription factor expression with branches
- Identify regulatory motifs in accessible chromatin
- Prioritize candidate regulators
Validation:
- Compare with known lineage markers
- Validate predictions using orthogonal methods
- Assess robustness through parameter perturbation
Table 2: Research Reagent Solutions for Multimodal Single-Cell Studies
| Reagent/Kit | Function | Application in Differentiation Studies |
|---|---|---|
| Single-Cell Multimale ATAC + Gene Expression Kit | Simultaneous profiling of chromatin accessibility and gene expression | Identifies coordinated changes in gene regulation during differentiation |
| Viability Staining Dyes | Distinguish live from dead cells | Ensures high-quality data by excluding compromised cells |
| Nuclei Isolation Buffers | Extract intact nuclei for ATAC-seq | Enables chromatin accessibility profiling from difficult tissues |
| Cell Hashing Antibodies | Multiplex samples by labeling with barcoded antibodies | Increases throughput and reduces batch effects in time-course studies |
| Single-Cell Barcoding Beads | Label individual cells with unique barcodes | Enables sequencing of thousands of cells in parallel |
Application Notes: Implementing PHLOWER for Kidney Organoid Differentiation
Case Study: Kidney Organoid Differentiation Analysis
Background: Kidney organoids derived from human pluripotent stem cells represent a powerful model for studying renal development and disease. However, these organoids often contain off-target cell types and exhibit heterogeneity that limits their utility. Applying PHLOWER to kidney organoid multimodal data enables detailed characterization of differentiation trajectories and identification of factors driving off-target cell formation.
Experimental Design:
- Generate time-course multimodal data during organoid differentiation
- Collect samples at multiple time points for trajectory reconstruction
- Include spatial transcriptomics data when available
- Validate findings with functional assays
PHLOWER Implementation:
- Data Integration: Combine scRNA-seq and scATAC-seq data from day 0, 7, 14, 21, and 28 of organoid differentiation.
- Trajectory Inference: Apply PHLOWER to reconstruct the complete differentiation tree from pluripotent state to mature renal cell types.
- Branch Point Analysis: Identify critical decision points where nephron progenitor cells commit to different renal lineages.
- Regulator Identification: Discover transcription factors associated with branching events and off-target differentiation.
Key Findings:
- PHLOWER successfully reconstructed complex branching trajectories leading to podocytes, proximal tubules, and distal tubules
- Identified previously uncharacterized transitional states in nephron formation
- Revealed transcription factors driving off-target neural and muscle cells
- Provided insights for improving differentiation protocols to reduce off-target populations
Protocol 3: Validation of Computational Predictions
Objective: Experimentally validate trajectory inferences and regulator identifications from PHLOWER analysis.
Materials:
- Validated antibodies for candidate proteins
- CRISPR/Cas9 system for gene perturbation
- qPCR reagents
- Immunofluorescence staining reagents
- Flow cytometry equipment
Procedure:
- Spatial Validation:
- Perform immunofluorescence for predicted markers of transitional states
- Correlate protein expression with computational predictions
- Confirm spatial organization of differentiation trajectories
Functional Validation:
- Knock out predicted transcription factors using CRISPR/Cas9
- Assess impact on differentiation by flow cytometry
- Measure expression of lineage markers by qPCR
Dynamic Validation:
- Perform live imaging of reporter cell lines
- Track cell fate decisions in real-time
- Compare with computational trajectory predictions
Interpretation Guidelines:
- Consistent spatial expression patterns support trajectory predictions
- Perturbation of regulators should alter branching proportions
- Live imaging should reveal transitions through predicted states
Data Analysis and Interpretation Guidelines
Quantitative Benchmarking of Trajectory Methods
Performance Metrics: When evaluating PHLOWER against other trajectory inference methods, researchers should employ multiple quantitative metrics to assess different aspects of performance. Key metrics include:
- Accuracy of Branch Order: Measures how well the inferred tree topology matches known lineage relationships
- Pseudotemporal Ordering Accuracy: Quantifies correlation with known temporal sequences
- Cell State Separation: Assesses clarity of distinction between terminal states
- Robustness to Noise: Evaluates stability of results with varying data quality
Table 3: Benchmarking Results for Trajectory Inference Methods
| Method | Multimodal Data Support | Complex Branching Capacity | Regulator Identification | Computational Efficiency |
|---|---|---|---|---|
| PHLOWER | Native support for multiple modalities | Excellent for multi-branching trees | Directly identifies regulators | Moderate to high |
| PAGA | Limited to unimodal or integrated data | Handles moderate complexity | Requires additional analysis | High |
| Slingshot | Unimodal only | Limited to simple trajectories | Not supported | High |
| Monocle 3 | Limited multimodal integration | Good for complex trees | Partial support | Moderate |
Interpretation of PHLOWER Results
Differentiation Tree Analysis: When interpreting PHLOWER output, researchers should focus on several key aspects of the reconstructed differentiation tree:
- Branch Point Significance: Assess the robustness of inferred branch points through bootstrapping or other resampling methods
- Transition States: Identify cells occupying positions between stable states, which may represent important transitional populations
- Trajectory Confidence: Evaluate the certainty of cell assignments to specific paths in the differentiation tree
- Regulator Validation: Corroborate predicted transcriptional regulators with existing literature and experimental data
Common Artifacts and Solutions:
- Discontinuous Trajectories: May result from insufficient sampling of intermediate states; consider increasing time points or cell numbers
- Spurious Branches: Can arise from batch effects; ensure proper data integration and normalization
- Over-connection: May occur with overly permissive parameters; adjust neighborhood size and complexity constraints
Integration with Functional Data: For maximum biological insight, PHLOWER results should be integrated with additional functional data:
- Gene Ontology Analysis: Enrichment of biological processes along differentiation branches
- Regulatory Network Inference: Construction of gene regulatory networks driving fate decisions
- Disease Association Mapping: Overlap of differentiation paths with disease-associated genetic variants
- Drug Response Correlation: Association of differentiation states with therapeutic sensitivity
These integrative analyses transform the computational outputs of PHLOWER into biologically actionable insights, enabling researchers to not only map differentiation pathways but also understand their functional significance in development, disease, and therapeutic contexts.
How PHLOWER Works: From Mathematical Principles to Practical Implementation
Application Notes: Core Algorithm and Workflow
The PHLOWER algorithm represents a significant advancement in computational trajectory analysis for single-cell data. It leverages the harmonic component of the Hodge Laplacian (HL) decomposition on simplicial complexes (SCs) to infer complex, multi-branching cell differentiation trajectories from multimodal single-cell sequencing data [1] [9].
The foundational insight of PHLOWER is that cellular differentiation processes can be mathematically represented and inferred by analyzing the "holes" in the topological structure of the data. The algorithm transforms single-cell data into a higher-order network (a simplicial complex), applies spectral decomposition to the Hodge Laplacian operator, and uses the resulting harmonic components to create embeddings that directly represent cell differentiation events and paths [1].
Table 1: Key Mathematical Components of the PHLOWER Algorithm
| Component | Mathematical Role | Biological Interpretation |
|---|---|---|
| Simplicial Complex (SC) | Higher-order generalization of a graph consisting of nodes (0-simplices), edges (1-simplices), and triangles (2-simplices) [1]. | Represents the topological structure of the single-cell data landscape. |
| Hodge Laplacian (HL) | A matrix operator generalizing the graph Laplacian, capturing relationships between edges via nodes and triangular faces [1] [9]. | Encodes the potential dynamics and flow of cell state transitions. |
| Hodge Decomposition | Decomposes edge flows into three orthogonal components: gradient-free, curl-free, and harmonic [1] [9]. | Separates different types of cellular state changes and progression. |
| Harmonic Component | The part of the decomposition associated with the null space of the HL, linked to holes in the SC [1]. | Reveals the presence and structure of major cell differentiation trajectories. |
Experimental Protocols
Protocol 1: Differentiation Tree Inference with PHLOWER
This protocol details the primary workflow for inferring a cell differentiation tree from single-cell multimodal data [1] [9].
Input: Preprocessed single-cell multimodal data (e.g., scRNA-seq + scATAC-seq).
Procedure:
- Graph Construction: Represent the single-cell data as a k-nearest neighbor (k-NN) graph, where each node corresponds to a single cell.
- Pseudotime Estimation: Calculate pseudotime for each cell using the graph Laplacian, similar to methods like Diffusion Maps. This step orders cells along a putative developmental timeline and identifies progenitor cells (low pseudotime) and terminal differentiated cells (high pseudotime) [1].
- Simplicial Complex Formation: Perform a Delaunay triangulation on the graph. To explicitly capture branching trajectories, add "dummy" edges that connect terminal cells back to progenitor cells. Each main trajectory in the graph will thereby form a "hole" in the simplicial complex structure [1] [9].
- Hodge Laplacian Decomposition: Compute the discrete Hodge Laplacian on the constructed simplicial complex. Perform a spectral decomposition of this operator to obtain its harmonic eigenvectors [1] [9].
- Trajectory Embedding: a. Path Sampling: Sample paths (edge flows) within the simplicial complex by executing a preference random walk on the k-NN graph, following edges with positive divergence (indicating increasing pseudotime). Each path represents a potential cell differentiation trajectory [9]. b. Harmonic Projection: Project these sampled paths onto the harmonic space derived from the Hodge decomposition. This creates a trajectory embedding, where each point represents a specific differentiation trajectory [1] [9].
- Tree Reconstruction: Apply a clustering analysis (e.g., DBSCAN) on the cumulative trajectory embedding space to group paths into distinct trajectory groups. Reconstruct the final differentiation tree by examining the evolution of these trajectories over pseudotime and identifying branching points. A branching point is detected when the distance between edges within bins, defined by the formula
d(i, j, k) = b_i^k/σ_i^k - b_j^k/σ_j^k, falls below a thresholdσ, whereb_i^kis the average edge coordinate per bin andσ_i^kis the average distance between edges in a bin [9].
Output: A complex, multi-branching cell differentiation tree.
Protocol 2: Benchmarking PHLOWER Against Competing Methods
This protocol outlines the benchmarking procedure used to validate PHLOWER's performance against other trajectory inference methods, as described in the original publication [1].
Input: Simulated datasets and real single-cell RNA-sequencing (scRNA-seq) datasets with known or well-established tree structures.
Procedure:
- Dataset Curation: a. Simulated Data: Utilize the diffusion-limited aggregation (DLA) model to generate ten complex differentiation tree datasets containing 5 to 18 total branches [1]. b. Real scRNA-seq Data: Select 33 single-rooted tree-structured datasets from the Dynverse benchmarking repository, ensuring each has at least three branches [1].
- Method Execution: Run PHLOWER and competing trajectory inference methods (e.g., PAGA tree, Monocle3, Slingshot, TSCAN) on the curated datasets [1].
- Performance Evaluation: Use the Dynverse framework to calculate the following metrics for each method and dataset combination [1]:
- Hamming–Ipsen–Mikhailov (HIM): Quantifies the similarity between the inferred and true tree topologies.
- Correlation: Measures the accuracy of cell placement within a branch.
- F1 Branches: Evaluates the correct allocation of cells to branches.
- F1 Milestones: Assesses the correct allocation of cells to branching points.
- Overall Accuracy: The average of the four above metrics.
- Computational Resource Assessment: Record the runtime and memory consumption for each method on datasets of varying sizes (e.g., pancreas progenitor with ~3,700 cells and neurogenesis with ~18,000 cells). Evaluate a downsampling strategy for PHLOWER to improve time efficiency [1].
Output: A comprehensive benchmarking report detailing the accuracy and computational efficiency of each method.
Table 2: Key Benchmarking Metrics and PHLOWER's Performance Profile
| Evaluation Metric | What It Measures | PHLOWER's Performance |
|---|---|---|
| HIM (Tree Similarity) | Topological accuracy of the inferred tree [1]. | Top performer on both simulated and real data [1]. |
| Cell-Branch Correlation | Correct ordering of cells along a branch [1]. | Top performer on both simulated and real data [1]. |
| F1 Branches Score | Accuracy of assigning cells to the correct branch [1]. | Top performer on simulated data; among the best on real data [1]. |
| Computational Demand | Runtime and memory usage [1]. | 0.5-12 hours and 12-40 GB for 3.7k-18k cells. Downsampling to 30% of cells greatly improves speed and memory use [1]. |
Protocol 3: Identification of Branch-Specific Transcriptional Regulators
This protocol describes how PHLOWER leverages multimodal data to identify transcription factors (TFs) associated with specific differentiation branches [1].
Input: A fitted PHLOWER model and matched single-cell multimodal data (e.g., gene expression from RNA-seq and TF activity from ATAC-seq analyzed with tools like ChromVAR).
Procedure:
- Trajectory Annotation: Based on the inferred tree, identify the populations of cells that constitute the end points of distinct branches.
- Differential Analysis: Perform statistical tests to identify genes and TFs that are differentially expressed or active between the terminal cell states of different branches.
- Regulator Validation: For candidate TFs, ensure there is concordance between their gene expression level and their inferred activity (e.g., from ChromVAR analysis). This step confirms that the TF is not only expressed but also functionally active in driving the branch-specific program [1].
- Functional Linkage: The identified TFs are implicated as key regulators of the transcriptional programs defining the specific cell fates at the end of each branch [1].
Output: A list of high-confidence, branch-specific transcriptional regulators.
The Scientist's Toolkit
Table 3: Essential Research Reagents and Computational Tools
| Item / Resource | Function / Role in the Workflow |
|---|---|
| Single-cell Multimodal Data (e.g., from 10x Genomics) | Primary input data measuring both gene expression and chromatin accessibility in the same cell [1]. |
| Kidney Organoid / Pancreas Progenitor Datasets | Example real-world biological systems used for validating PHLOWER's performance [1]. |
| Delaunay Triangulation Algorithm | Computational method for constructing the simplicial complex from the single-cell graph [1] [9]. |
| ChromVAR | Software tool for inferring transcription factor activity from single-cell ATAC-seq data [1]. |
| Dynverse Framework | A benchmarking platform providing datasets and standardized metrics for evaluating trajectory inference methods [1]. |
| DBSCAN Clustering | Algorithm used within PHLOWER to group sampled paths into distinct trajectories in the embedding space [9]. |
Cellular differentiation is a fundamental process in development and disease, during which a cell undergoes changes in its chromatin and gene expression programs to acquire specialized functions. A key challenge in studying this process is that single-cell sequencing technologies, while powerful, require the dissociation of cells, making it impossible to experimentally track the differentiation of an individual cell over time. Computational trajectory inference has therefore become an indispensable tool for reconstructing these temporal processes from static single-cell snapshots [1].
The emergence of single-cell multimodal sequencing, which can measure both the full transcriptome and genome-wide chromatin accessibility from the same cell, provides a unique resource to understand the interplay between chromatin state, transcription factor (TF) binding, and gene expression changes during differentiation [1]. However, existing computational approaches have primarily been applied to study differentiation trees with limited complexity (typically 3-9 branches), with no comprehensive benchmarking on complex, multi-branching structures. The PHLOWER method (decomposition of the Hodge Laplacian for inferring trajectories from flows of cell differentiation) was developed specifically to address this challenge by leveraging the harmonic component of the Hodge decomposition on simplicial complexes to infer trajectory embeddings from single-cell multimodal data [1] [3].
The PHLOWER Method: Theoretical Foundation and Workflow
Mathematical Underpinnings: From Graph Laplacian to Hodge Laplacian
PHLOWER represents a significant advancement in trajectory inference through its application of higher-order topological data analysis. While traditional methods often rely on the Graph Laplacian (a zero-order Hodge Laplacian), where samples are encoded as vertices and distances as edge weights, PHLOWER extends this concept by representing single-cell data as a simplicial complex (SC) [1].
A simplicial complex is a generalization of a graph that incorporates not only nodes (0-simplices) and edges (1-simplices), but also triangles (2-simplices) and other higher-order structures. The spectral decomposition of the Hodge Laplacian on an SC decomposes edge flows into three fundamental components: gradient-free, curl-free, and harmonic components. PHLOWER specifically focuses on the harmonic eigenvectors of the Hodge Laplacian, which are associated with "holes" in the simplicial complex that correspond to cell differentiation tree branches in the gene expression space [1].
Theoretical guarantees ensure that the behavior of the Hodge Laplacian on a simplicial complex converges to the Hodge Laplacian on manifolds in the limit, providing a robust mathematical foundation for the method [1].
Computational Workflow
The PHLOWER algorithm implements a multi-step workflow to transform raw single-cell data into complex differentiation trajectories:
Graph Representation and Pseudotime Estimation: PHLOWER first creates a graph representation of the single-cell data and uses the graph Laplacian to estimate pseudotime of cells and identify progenitor (low pseudotime) and terminal differentiated cells (high pseudotime), similar to established methods [1].
Simplicial Complex Construction: The directed branching process of differentiation is transformed into a simplicial complex through Delaunay triangulation, connecting terminal differentiated cells with progenitor cells. This connection creates a "hole" for every main trajectory in the graph [1].
Hodge Decomposition and Embedding: The algorithm performs a Hodge decomposition of the edge-flow space on the simplicial complex. The harmonic components of this decomposition provide two critical embeddings:
- Edge-level embeddings: Each point represents a cell differentiation event
- Trajectory embeddings: Each point represents a cell differentiation path [1]
Trajectory Identification: These natural representations of cell differentiation facilitate the estimation of underlying differentiation trees by delineating major differentiation trajectories [1].
The following diagram illustrates the core computational workflow of the PHLOWER method:
Experimental Protocols and Implementation
Dataset Preparation and Preprocessing
Successful application of PHLOWER requires appropriate dataset preparation. Multiple publicly available single-cell datasets can serve as ideal starting points:
Human Pancreas Datasets:
- Muraro et al. (2016): Single-cell CEL-seq2 data generated using a customized automated platform that combines FACS, robotics, and the CEL-Seq2 protocol to obtain transcriptomes of thousands of single pancreatic cells from four deceased organ donors [10].
- Segerstolpe et al. (2016): Single-cell RNA-seq dataset of human pancreatic cells from patients with type 2 diabetes and healthy controls. Single cells were prepared using Smart-seq2 protocol and sequenced on an Illumina HiSeq 2000 [10].
Tabula Muris Dataset:
- A comprehensive resource containing almost 100,000 cells across 20 different mouse tissues/organs, combining high-throughput but low-coverage 10X data with lower throughput but high-coverage FACS-sorted cells plus Smartseq2 [10].
- Download via figshare using DOI: 10.6084/m9.figshare.5715040 for FACS/Smartseq2 and 10.6084/m9.figshare.5715025 for 10X data [10].
Data Preprocessing Protocol:
- Quality Control: Filter cells based on mitochondrial content, number of detected genes, and total counts
- Normalization: Normalize counts across cells to account for varying sequencing depth
- Feature Selection: Identify highly variable genes for downstream analysis
- Dimension Reduction: Perform initial PCA to reduce dimensionality
- Batch Effect Correction: Apply appropriate methods (e.g., Harmony, BBKNN) if multiple batches are present
PHLOWER Implementation Protocol
Software Requirements and Installation:
- Python 3.8 or higher with scientific computing stack (NumPy, SciPy, pandas)
- Specialized topology packages (available via PyPI)
- Minimum 12-40 GB RAM recommended for standard datasets [1]
Execution Steps:
- Data Input: Load preprocessed single-cell data (expression matrix, chromatin accessibility data if multimodal)
- Parameter Configuration:
- Set k-nearest neighbors for graph construction (default: 30)
- Configure pseudotime estimation method (default: diffusion maps)
- Define simplicial complex parameters (Delaunay triangulation thresholds)
- Algorithm Execution:
- Construct cell-to-cell graph
- Estimate pseudotime and identify progenitor/terminal states
- Build simplicial complex with Delaunay triangulation
- Compute Hodge Laplacian and perform spectral decomposition
- Extract harmonic components for trajectory embedding
- Trajectory Extraction:
- Identify major branches from harmonic embeddings
- Assign cells to specific differentiation paths
- Reconstruct complete differentiation tree
Computational Considerations: For the pancreas progenitor dataset (~3,700 cells), PHLOWER requires 0.5-12 hours and 12-40 GB of memory on a standard desktop. A time-efficient variant using cell downsampling (30% of cells) provides 8.6× faster processing and uses one-sixth of the memory while still inferring accurate differentiation trees [1].
Benchmarking and Validation Protocol
To validate PHLOWER performance against competing methods, implement the following benchmarking protocol:
Datasets for Benchmarking:
- Simulated Data: Use diffusion-limited aggregation (DLA) trees to simulate ten complex differentiation tree datasets with 5 to 18 total branches [1]
- Real scRNA-seq Data: Select 33 scRNA-seq datasets from Dynverse containing single-rooted tree structures with at least three branches [1]
Comparative Methods: Include PAGA tree, Monocle3, cellTree, pCreode, Slice, RaceID, Slingshot, TSCAN, MST, Elpigraph, and STREAM in the benchmarking [1].
Evaluation Metrics (using Dynverse framework):
- Tree structure similarity (Hamming–Ipsen–Mikhailov, HIM)
- Location of cells within a branch (correlation)
- Cell allocation to branches (F1 branches)
- Cell allocation to branching points (F1 milestones)
- Overall accuracy (average of the four previous metrics) [1]
Table 1: Benchmarking Results of PHLOWER Against Competing Methods
| Method | Simulated Data Accuracy | Real scRNA-seq Data Accuracy | Complex Tree Performance | Computational Efficiency |
|---|---|---|---|---|
| PHLOWER | Highest | Highest | Excellent | Moderate (0.5-12 hours for 3,700 cells) |
| PAGA | High | High | Good | High |
| Monocle3 | High | High | Good | Moderate |
| RaceID | High | Moderate | Moderate | Moderate |
| Slingshot | Moderate | Moderate | Poor | High |
| TSCAN | Moderate | Moderate | Poor | High |
Research Reagent Solutions and Essential Materials
Successful implementation of single-cell trajectory analysis requires both computational tools and appropriate experimental reagents. The following table details essential materials and their functions:
Table 2: Essential Research Reagents and Computational Tools for Single-Cell Trajectory Analysis
| Reagent/Tool | Function | Example Applications | Key Features |
|---|---|---|---|
| Smart-seq2 | High-coverage full-length scRNA-seq protocol | Deng dataset (mouse preimplantation development) [10] | Full transcript coverage, detects isoform usage |
| 10X Genomics | High-throughput droplet-based scRNA-seq | Tabula Muris droplet data [10] | High cell throughput, UMI-based counting |
| CEL-seq2 | Automated plate-based scRNA-seq | Muraro pancreas dataset [10] | Low PCR duplication rates, well-based |
| Fluidigm C1 | Microfluidic scRNA-seq platform | Tung iPSC dataset [10] | Integrated cell capture and processing |
| PHLOWER | Trajectory inference from multimodal data | Kidney organoid analysis, hematopoiesis [1] [3] | Handles complex branching, uses Hodge Laplacian |
| Trailmaker | User-friendly scRNA-seq analysis platform | Analysis of Parse Biosciences Evercode WT data [11] | No coding required, Monocle3 trajectory analysis |
| Dynverse | Benchmarking platform for trajectory methods | Standardized evaluation of 33 datasets [1] | Consistent metrics, multiple method comparison |
Advanced Applications and Biological Insights
Case Study: Hematopoietic Differentiation Hierarchy
The power of single-cell analysis for revealing complex differentiation pathways is exemplified by studies of the mouse hematopoietic system. Through highly multiplexed single-cell qPCR analysis of 280 cell surface markers across more than 1500 single cells, researchers have reconstructed the hematopoietic stem cell (HSC) differentiation hierarchy, challenging the conventional binary commitment model [12].
This approach revealed that the conventional view of lymphoid versus myeloid bifurcation as the first lineage specification event may be oversimplified. Instead, the data suggested that lymphomyeloid lineage commitment may occur upstream of the separation of common lymphoid progenitors (CLP) and common myeloid progenitors (CMP), demonstrating how comprehensive single-cell analysis can reshape fundamental biological models [12].
The following diagram illustrates the complex branching structure revealed by such single-cell analyses of hematopoietic differentiation:
Kidney Organoid Analysis and Transcription Factor Identification
In addition to reconstructing differentiation trajectories, PHLOWER enables the identification of transcription factors regulating specific differentiation paths. Application of PHLOWER to kidney organoid multimodal and spatial single-cell data demonstrated its power not only in inferring complex trees but also in identifying transcription factors regulating off-target cells in kidney organoids [1] [3].
This capability to link trajectory structure with regulatory mechanism identification makes PHLOWER particularly valuable for developmental biology and disease modeling applications, where understanding both the "path" and the "drivers" of differentiation is crucial.
Technical Considerations and Optimization Strategies
Visualization Principles for Biological Pathways
Effective visualization of complex differentiation trajectories is essential for interpretation and communication. The following design principles enhance clarity and interpretability:
Optimize Flow of Information: Arrange elements in logical, unidirectional pathways (left-to-right or top-to-bottom) to guide the viewer through the differentiation process [13].
Color and Contrast: Utilize color saturation and contrast to draw attention to key elements. Ensure sufficient contrast for colorblind accessibility by explicitly setting text color (fontcolor) to have high contrast against node background colors (fillcolor) [13] [14].
Zooming to Correct Scale: Focus viewer attention by zooming in on key information at the appropriate scale, particularly important when representing processes from cellular to molecular levels [13].
Consistent Lines and Arrows: Maintain consistent arrow styles and line weights to unify illustrations and clearly communicate differentiation directions and relationships [13].
Performance Optimization for Large Datasets
For analysis of large single-cell datasets (≥10,000 cells), implement these optimization strategies:
- Cell Downsampling: Use 30% of cells for 8.6× faster processing and one-sixth memory usage while maintaining trajectory accuracy [1]
- Sparse Matrix Operations: Leverage sparse matrix representations for memory-efficient computation [10]
- Approximate Nearest Neighbors: Implement approximate algorithms for graph construction in large datasets
- Parallel Processing: Distribute computational loads across multiple cores for Hodge Laplacian decomposition
PHLOWER represents a significant advancement in computational trajectory inference by addressing the critical challenge of predicting complex, multi-branching trees from multimodal single-cell data. Through its novel application of Hodge decomposition on simplicial complexes, PHLOWER enables researchers to move beyond simple bifurcating models to capture the true complexity of cellular differentiation hierarchies.
The method's superior performance in benchmarking studies, particularly for complex tree structures, combined with its ability to identify key transcriptional regulators, makes it particularly valuable for both basic developmental biology and applied drug development research. As single-cell multimodal technologies continue to evolve, approaches like PHLOWER will be increasingly essential for extracting the full biological insights from these rich datasets.
Trajectory inference is a computational technique used in single-cell transcriptomics to determine the pattern of a dynamic process experienced by cells and then arrange cells based on their progression through the process [15]. This approach characterizes dynamic processes such as cell differentiation, the cell cycle, or response to external stimuli by placing cells along a continuous path representing the evolution of the process, rather than dividing them into discrete clusters [15]. The positioning of cells along the trajectory is quantified as "pseudotime," which reflects the relative activity or progression of the underlying biological process [16]. This protocol details a comprehensive workflow from pseudotime estimation to final trajectory embedding, contextualized within broader research on the PHLOWER method, which leverages harmonic components of the Hodge decomposition on simplicial complexes to infer flow embeddings from single-cell multimodal data [4].
Key Concepts and Biological Significance
Pseudotime is a fundamental scalar metric that assigns a continuous value to each cell, quantifying its progression along an inferred developmental path [4]. It is typically derived from the projection of a cell's expression profile onto a parameterized trajectory curve [4]. Unlike real chronological time, pseudotime describes the relative transition of cellular states, with cells having larger pseudotime values considered more "advanced" in the process [16]. For branched trajectories, multiple pseudotime values are typically assigned, one per path through the trajectory, and these values are not usually comparable across paths [16].
Trajectory inference methods rely on several core assumptions: (1) the dataset captures a continuous biological process where gene expression changes gradually across states; (2) cells represent independent, asynchronous samples drawn from this trajectory at different progression points; (3) an underlying low-dimensional manifold structure exists in the data; and (4) sufficient sampling density across trajectory states exists to avoid gaps that could distort reconstruction [4]. Violations of these assumptions can introduce artifacts, such as erroneous linearization of cyclic processes, leading to biologically implausible trajectories [4].
The following workflow outlines the major stages from data preprocessing through trajectory interpretation, with particular emphasis on methodologies compatible with the PHLOWER framework.
Workflow Diagram
Materials and Methods
Research Reagent Solutions
Table 1: Essential research reagents and computational tools for trajectory inference
| Category | Item/Software | Function | Implementation |
|---|---|---|---|
| Data Processing | Seurat/SingleCellExperiment | Data container and preprocessing | R/Python |
| SCANPY | Single-cell analysis in Python | Python | |
| scran | Scalable normalization | R | |
| Dimensionality Reduction | PCA | Linear dimensionality reduction | R/Python |
| UMAP | Nonlinear visualization | R/Python | |
| Diffusion Maps | Manifold learning for trajectories | R/Python | |
| Trajectory Inference | PHLOWER | Flow embedding from multimodal data | [4] |
| Monocle 3 | Large-scale trajectory inference | R | |
| Slingshot | Cluster-based principal curves | R | |
| PAGA | Graph abstraction with topology preservation | Python | |
| TSCAN | MST-based trajectory reconstruction | R | |
| Downstream Analysis | tradeSeq | Differential expression along paths | R |
| CellRank | Multi-view fate prediction | Python | |
| velocyto | RNA velocity estimation | Python/R |
Detailed Protocol
Data Preprocessing and Quality Control
Begin with raw single-cell RNA sequencing count data. Perform quality control to remove low-quality cells based on metrics including total counts, number of detected genes, and mitochondrial percentage. Normalize data using method such as scran pooling-based size factors or SCANPY's default normalization. Select highly variable genes (HVGs) for downstream analysis, typically 2,000-5,000 features that exhibit the highest cell-to-cell variation.
Dimensionality Reduction and Clustering
Apply principal component analysis (PCA) to the normalized and scaled HVG matrix to reduce dimensionality while preserving biological variance. Use the elbow plot of variance explained to determine the number of significant PCs to retain for downstream analysis. Perform clustering using graph-based methods (Louvain/Leiden) in the PC space to identify discrete cell states or types. These clusters will serve as nodes for trajectory construction in subsequent steps.
Trajectory Inference Methods
Different trajectory inference methods employ distinct strategies for reconstructing cellular paths, each with specific strengths for particular biological contexts and dataset characteristics.
PHLOWER Method Implementation: The PHLOWER method leverages the harmonic component of the Hodge decomposition on simplicial complexes to infer flow embeddings from single-cell multimodal data [4]. This approach has been evaluated in comprehensive benchmarking datasets with large cell differentiation trees and was used to dissect regulatory programs controlling kidney organoid cell differentiation [4]. Implement PHLOWER following these steps:
- Construct a simplicial complex representation from multimodal single-cell data
- Apply Hodge decomposition to separate gradient, harmonic, and curl components
- Extract the harmonic component representing the trajectory flow
- Project cells onto the flow embedding to obtain pseudotime values
Slingshot Implementation: Slingshot uses cluster labels as input and orders these clusters into lineages by constructing a minimum spanning tree (MST) [17] [15]. Paths through the tree are smoothed by fitting simultaneous principal curves, and a cell's pseudotime value is determined by its projection onto one or more of these curves [15]. The algorithm is implemented as follows:
- Input cluster labels and low-dimensional representation (PCA, UMAP)
- Construct MST on cluster centroids to determine global lineage structure
- Identify starting cluster (manually specified or automatically detected)
- Fit simultaneous principal curves through each lineage
- Project cells to nearest curve to calculate pseudotime
Monocle 3 Implementation: Monocle 3 uses reversed graph embedding to reconstruct trajectories and supports complex topologies including cycles and multiple branches [17]. The workflow includes:
- Project high-dimensional data to lower dimensions using UMAP
- Cluster cells using Louvain/Leiden algorithm in reduced space
- Construct graph using variant of SimplePPT algorithm
- Assign pseudotime by projecting samples onto trajectory and computing distance from root node
PAGA Implementation: Partition-based Graph Abstraction (PAGA) creates a graph-like map of single-cell data that preserves both continuous and disconnected structures [17]. Implementation steps:
- Compute k-nearest neighbor graph in PCA space
- Apply Louvain/Leiden clustering to identify partitions
- Build PAGA graph connecting partitions based on statistical significance of connectivity
- Generate pseudotime by computing shortest paths through PAGA graph from root node
Pseudotime Calculation and Visualization
After trajectory inference, calculate pseudotime values for each cell. For PHLOWER, pseudotime is derived directly from position along the harmonic flow embedding [4]. For other methods, pseudotime is typically computed as the distance along the trajectory from a user-specified root node representing the starting state [17]. Visualize results by plotting cells in a low-dimensional embedding (UMAP, t-SNE) colored by pseudotime values. For branched trajectories, create separate pseudotime values for each lineage.
Validation and Downstream Analysis
Validate trajectory results using methods such as self-consistency tests (subsampling stability) [17] and comparison to known marker genes. Perform differential expression analysis along pseudotime to identify genes associated with progression (e.g., using tradeSeq) [4]. Analyze branch points to detect genes associated with fate decisions using likelihood ratio tests. For PHLOWER specifically, leverage the harmonic nature of the embedding to identify regulatory programs controlling differentiation processes [4].
Method Comparison and Selection Criteria
Table 2: Comparative analysis of trajectory inference methods
| Method | Topology | Strengths | Limitations | Best Use Cases |
|---|---|---|---|---|
| PHLOWER | Complex trees | Multimodal integration, flow embedding | Method complexity | Kidney organoids, complex differentiation |
| Slingshot | Branching | Robust to noise, flexible clustering input | Requires pre-clustering | General differentiation studies |
| Monocle 3 | Complex | Scalable to millions of cells, multiple trajectories | Computationally intensive | Large datasets, complex topologies |
| PAGA | Disconnected/connected | Preserves disconnected clusters, interpretable graph | Coarse-grained structure | Heterogeneous samples with multiple lineages |
| TSCAN | Linear, branching | Fast, interpretable MST-based approach | Limited to tree structures | Initial exploratory analysis |
Applications in Drug Development and Disease Research
Trajectory inference has significant applications in pharmaceutical research and development. By revealing cellular transition states and differentiation pathways, these methods can identify novel drug targets at critical decision points in disease progression [4]. In oncology, trajectory analysis can reveal tumor heterogeneity and evolution, including resistance mechanisms [4]. For immunology, it can track immune cell activation and differentiation in response to therapies [4]. The PHLOWER method specifically has been applied to dissect regulatory programs controlling kidney organoid cell differentiation, demonstrating utility in tissue engineering and regenerative medicine applications [4].
Troubleshooting and Technical Considerations
Data Quality Issues: If trajectories appear fragmented or discontinuous, assess whether the data has sufficient cells capturing transition states. Increase cell numbers or consider using methods like PAGA that handle disconnected structures [17]. High technical noise can obscure biological trajectories; apply more stringent quality control or denoising techniques.
Method Selection: For datasets with expected complex trajectories (multiple branches, cycles), select methods like Monocle 3 or PHLOWER that support these topologies [17] [4]. For linear processes or initial exploration, TSCAN or Slingshot may be sufficient.
Validation Challenges: Always validate trajectories using known marker genes or experimental validation when possible. Be aware that extreme dimensionality reduction to 2D can induce significant distortion of high-dimensional relationships [18]. Use multiple visualizations and statistical assessments rather than relying solely on 2D embeddings.
PHLOWER-Specific Considerations: When applying PHLOWER, ensure multimodal data is properly integrated and normalized. The harmonic decomposition requires careful parameter tuning for simplicial complex construction. Refer to benchmarking studies for guidance on implementation details [4].
Kidney organoids, three-dimensional structures derived from pluripotent stem cells (PSCs), have emerged as powerful in vitro models that recapitulate key aspects of kidney development, structure, and function [19]. These organoids contain self-organized nephron-like structures, including glomerular, proximal tubular, and distal tubular segments, providing a physiologically relevant platform for studying human kidney biology and disease [19]. The complexity of kidney organoids presents both an opportunity and a challenge for comprehensive analysis. Recent advances in single-cell multimodal technologies now enable researchers to simultaneously measure multiple molecular modalities—such as gene expression and chromatin accessibility—from the same cell, creating rich datasets that capture the intricate processes of cell differentiation and tissue organization [1].
The PHLOWER (decomposition of the Hodge Laplacian for inferring trajectories from flows of cell differentiation) method represents a significant computational advancement for analyzing such complex datasets [1] [3]. This approach leverages the harmonic component of the Hodge decomposition on simplicial complexes to infer trajectory embeddings from single-cell multimodal data, enabling the reconstruction of complex, multi-branching cell differentiation trees [1]. When applied to kidney organoid research, PHLOWER provides a powerful framework for deciphering the developmental trajectories and transcriptional regulators that guide nephrogenesis, offering particular utility for identifying off-target cells and optimizing differentiation protocols [1].
This application note details experimental and computational protocols for generating and analyzing kidney organoid multimodal and spatial data, with specific emphasis on integration with the PHLOWER method for inferring cell differentiation trajectories.
Kidney Organoid Generation and Multimodal Data Production
Protocol for Kidney Organoid Differentiation from Pluripotent Stem Cells
The generation of kidney organoids in vitro follows a stepwise protocol that recapitulates developmental principles, directing PSCs through primitive streak, intermediate mesoderm, and metanephric mesenchyme stages [19].
Key Materials:
- Human induced pluripotent stem cells (iPSCs) or embryonic stem cells (ESCs)
- CHIR99021 (GSK3β inhibitor)
- Fibroblast growth factor 9 (FGF9)
- Bone morphogenetic protein 7 (BMP7) - Optional, protocol-dependent
- Low-adhesion culture plates for 3D spheroid formation
Detailed Methodology:
Primitive Streak Induction: Treat PSCs with CHIR99021 to activate canonical WNT signaling, driving differentiation toward posterior primitive streak identity [19]. Concentration and timing of CHIR99021 treatment require optimization for specific cell lines.
Intermediate Mesoderm Patterning: After 2-4 days of WNT activation, pattern the primitive streak cells toward intermediate mesoderm using FGF9, with some protocols additionally incorporating BMP7 [19].
3D Aggregation and Nephrogenesis: Dissociate the differentiated cells and transfer to low-adhesion plates to promote 3D spheroid formation. Continue FGF9 exposure to support the maintenance and expansion of nephron progenitor populations, which will subsequently undergo mesenchymal-to-epithelial transition (MET) to form nephron-like structures [19] [1].
Maturation: Culture organoids for 10-21 days to allow development of segmented nephron structures. More advanced maturation can be achieved through dynamic culture systems (e.g., spinning bioreactors) or transplantation into immunodeficient mice to promote vascularization [19].
Quality Control Assessment:
- Structural Validation: Immunofluorescence staining for characteristic markers:
- Podocytes: WT1, NPHS1 (nephrin), PODXL (podocalyxin)
- Proximal Tubules: LTL (lotus tetragonolobus lectin), SLC3A1, CUBN
- Distal Tubules: ECAD (E-cadherin), SLC12A1 [19]
- Functional Assessment: Evaluate proximal tubule function through albumin uptake assays [19].
Protocol for Generating Multimodal Single-Cell and Spatial Data
Experimental Workflow for Multimodal Single-Cell Analysis:
- Single-Cell Multimodal Sequencing: Process kidney organoids at relevant timepoints (e.g., days 7, 10, 14, 21) using a multimodal single-cell technology such as 10X Genomics Multiome (ATAC + Gene Expression) or CITE-seq.
- Spatial Transcriptomics: For select timepoints, process adjacent organoid samples using spatial transcriptomic technologies such as 10X Genomics Visium, STARmap, MERFISH, or seqFISH to preserve spatial context [20].
- Library Preparation and Sequencing: Follow manufacturer protocols for library preparation and perform high-throughput sequencing.
Computational Preprocessing Pipeline:
- Data Demultiplexing and Alignment: Use platform-specific tools (e.g., Cell Ranger ARC, Space Ranger) to demultiplex raw sequencing data and align reads to the reference genome.
- Quality Control and Filtering:
- Remove cells with low unique molecular identifier (UMI) counts, high mitochondrial gene percentage, or low peak-to-fragment ratio (for ATAC data).
- Retain cells passing quality thresholds for subsequent analysis.
- Modality Integration: Use tools like Seurat (R) or Scanpy/Squidpy (Python) to create a unified object containing both gene expression and chromatin accessibility measurements from the same cells [20].
Table 1: Single-Cell Multimodal Sequencing Technologies
| Technology | Measured Modalities | Key Applications in Kidney Organoids | Resolution |
|---|---|---|---|
| 10X Multiome | Gene Expression + Chromatin Accessibility (ATAC-seq) | Linking regulatory landscapes to transcriptional outputs during nephrogenesis | Single-cell |
| CITE-seq | Gene Expression + Surface Proteins | Characterizing immune/stromal populations and cell surface phenotypes | Single-cell |
| SHARE-seq | Gene Expression + Chromatin Accessibility | Enhanced co-assay of gene regulation and open chromatin | Single-cell |
Table 2: Spatial Transcriptomic Platforms
| Platform | Technology Principle | Key Applications in Kidney Organoids | Resolution |
|---|---|---|---|
| 10X Visium | Spatially barcoded RNA capture spots | Mapping the spatial organization of nephron segments and stromal compartments | 55 μm |
| MERFISH | Multiplexed error-robust fluorescence in situ hybridization | High-plex imaging of predefined gene panels for cell typing | Single-cell |
| seqFISH | Sequential fluorescence in situ hybridization | In situ transcriptomics for spatial localization of cell types | Single-cell |
| STARmap | In situ sequencing | 3D intact-tissue spatial mapping of gene expression | Single-cell |
Computational Analysis with PHLOWER
Protocol for Trajectory Inference with PHLOWER
PHLOWER addresses the challenge of inferring complex, multi-branching differentiation trees from multimodal single-cell data by leveraging the Hodge Laplacian on simplicial complexes [1].
Prerequisites and Data Input:
- A processed single-cell multimodal dataset (e.g., after Seurat/Scanpy preprocessing) containing both gene expression and chromatin accessibility data.
- A computed cell embedding (e.g., PCA, scVI) representing the transcriptional state of each cell.
Detailed Computational Steps:
Pseudotime Estimation and Terminal/Progenitor Cell Identification:
Simplicial Complex Construction: Represent the single-cell data as a simplicial complex, generalizing a graph to include nodes (cells), edges (potential differentiation events), and triangles (higher-order relationships). PHLOWER performs a Delaunay triangulation, connecting terminal differentiated cells with progenitor cells to create topological "holes" corresponding to main differentiation trajectories [1].
Hodge Laplacian Decomposition: Perform spectral decomposition of the Hodge Laplacian on the simplicial complex to decompose edge flows into gradient-free, curl-free, and harmonic components [1].
Trajectory Embedding and Tree Inference: Extract the harmonic components to create edge-flow embeddings (representing cell differentiation events) and trajectory embeddings (representing cell differentiation paths). These natural representations facilitate the detection of complex differentiation trees [1].
Branch Annotation and Transcription Factor Analysis: Identify transcription factors regulating differentiation along each branch by correlating chromatin accessibility (ATAC-seq peaks) in regulatory regions with gene expression of potential regulator TFs along the inferred trajectories [1].
Workflow Diagram for Kidney Organoid Analysis with PHLOWER
Diagram 1: Integrated workflow for kidney organoid analysis with PHLOWER.
PHLOWER Performance Characteristics
Table 3: PHLOWER Benchmarking Performance Compared to Other Methods
| Method | Tree Structure Similarity (HIM) | Cell Location (Correlation) | Cell Branch Allocation (F1) | Computational Time (Large Dataset) |
|---|---|---|---|---|
| PHLOWER | Best Performance | Best Performance | Best Performance | 0.5-12 hours |
| PAGA | Runner-up | Moderate | Runner-up | Faster |
| Monocle3 | Runner-up | Runner-up | Runner-up | Moderate |
| Slingshot | Lower performance | Moderate | Lower performance | Fastest |
Note: Benchmarking data based on simulated datasets with 5-18 branches and 33 real scRNA-seq datasets from Dynverse [1].
Application Case Study: Identifying Off-Target Cells in Kidney Organoids
Experimental Protocol for Validating Off-Target Cell Populations
A key application of PHLOWER in kidney organoid research is the identification and characterization of "off-target" cell types—neuronal, muscle, or other non-renal lineages that may arise due to imperfect differentiation protocol specificity [1].
Analytical Workflow:
Data Generation and Preprocessing: Generate a single-cell multimodal dataset (RNA + ATAC) from day 21 kidney organoids using the described wet-lab protocols.
PHLOWER Trajectory Analysis: Run the PHLOWER pipeline to infer the complete differentiation tree. Branches that terminate in cell clusters expressing non-renal markers (e.g., neuronal: TUJ1, SOX10; muscle: ACTA2, MYOD1) are identified as off-target trajectories [1].
Spatial Validation: Correlate findings with spatial transcriptomic data from adjacent organoid sections to determine whether off-target cells form organized structures or are randomly distributed [20].
Regulator Identification: PHLOWER identifies transcription factors whose regulatory activity (inferred from combined ATAC+RNA data) is specifically enriched in the off-target branches [1].
Functional Validation: Design perturbation experiments (CRISPRi, small molecule inhibitors) targeting the identified transcription factors to suppress off-target differentiation in subsequent organoid differentiations.
Signaling Pathways in Kidney Organoid Development and Off-Target Differentiation
Diagram 2: Key signaling pathways in kidney organoid differentiation.
Table 4: Key Research Reagent Solutions for Kidney Organoid and Multimodal Studies
| Category | Item | Function/Application | Example |
|---|---|---|---|
| Differentiation Factors | CHIR99021 | GSK3β inhibitor; activates WNT signaling for primitive streak induction | Tocris Bioscience [#] |
| Recombinant FGF9 | Patterns intermediate mesoderm and supports nephron progenitor expansion | R&D Systems [#] | |
| Recombinant BMP7 | Aids in intermediate mesoderm patterning (protocol-dependent) | PeproTech [#] | |
| Cell Culture | Low-adhesion plates | Enables 3D spheroid formation for organoid self-organization | Corning Ultra-Low Attachment |
| Sequencing | Single-cell multimodal kit | Simultaneous measurement of gene expression and chromatin accessibility | 10X Genomics Multiome ATAC + Gene Expression |
| Spatial transcriptomics kit | Gene expression profiling with tissue context preservation | 10X Genomics Visium | |
| Antibodies | Anti-NPHS1 (nephrin) | Podocyte marker for immunofluorescence validation | Abcam [#] |
| Anti-LTL | Proximal tubule marker for immunofluorescence | Vector Laboratories [#] | |
| Anti-WT1 | Podocyte and nephron progenitor nuclear marker | Santa Cruz Biotechnology [#] | |
| Computational Tools | PHLOWER package | Inference of complex, multi-branching differentiation trees | Available on GitHub |
| SpacoR/Spaco | Spatially aware colorization for enhanced categorical data visualization | Available on GitHub [20] | |
| Seurat / Scanpy | Primary environments for single-cell and spatial data analysis | R/Bioconductor / Python |
The integration of kidney organoid biology with advanced computational methods like PHLOWER creates a powerful synergistic platform for developmental biology and disease modeling. This application note provides a comprehensive framework for generating high-quality kidney organoids, producing multimodal and spatial datasets, and applying the PHLOWER method to reconstruct complex differentiation trajectories and identify regulatory factors. This integrated approach accelerates the optimization of organoid differentiation protocols, enhances disease modeling fidelity, and provides deeper insights into the regulatory logic of human nephrogenesis, ultimately supporting drug screening and regenerative medicine applications.
Identifying Transcription Factors and Regulatory Mechanisms with PHLOWER
PHLOWER (decomposition of the Hodge Laplacian for inferring trajectories from flows of cell differentiation) represents a significant advancement in computational trajectory analysis for single-cell data. This method leverages the harmonic component of the Hodge decomposition on simplicial complexes to infer trajectory embeddings from single-cell multimodal data, enabling the estimation of underlying differentiation trees [1] [3]. Cellular differentiation is fundamentally regulated by transcription factors (TFs)—proteins that bind to regulatory DNA regions and orchestrate gene expression programs. Dissecting these key regulatory events is crucial for developing cellular reprogramming protocols, improving ex vivo differentiation in organoids, and understanding disease-related differentiation processes for therapeutic interventions [1]. PHLOWER addresses the critical challenge of predicting complex, multi-branching trees from multimodal data, which has remained an open problem in the field until now. By leveraging single-cell multimodal data, which captures both full expression programs and genome-wide open chromatin information, PHLOWER provides a unique resource to understand the interplay between chromatin, regulatory signals, and expression changes during cellular differentiation [1]. This application note details how researchers can utilize PHLOWER to identify key transcription factors and their regulatory mechanisms throughout cell differentiation processes.
Key Advantages and Performance Benchmarks
Quantitative Performance Evaluation
PHLOWER has demonstrated superior performance in comprehensive benchmarking against established trajectory inference methods. In evaluations using both simulated datasets and real single-cell RNA-sequencing (scRNA-seq) data, PHLOWER consistently outperformed competing approaches across multiple metrics [1].
Table 1: Performance Benchmarking of PHLOWER Against Competing Methods
| Evaluation Metric | Simulated Data Performance | Real scRNA-seq Data Performance | Top Performing Methods (Ranked) |
|---|---|---|---|
| Tree Topology Recovery | PHLOWER best performing [1] | PHLOWER best performing [1] | 1. PHLOWER2. PAGA3. RaceID/Monocle3 |
| Cell Location within Branches | PHLOWER best performing [1] | PHLOWER highest average scores [1] | 1. PHLOWER2. TSCAN/RaceID3. Monocle3/Slingshot |
| Cell Allocation to Branches | PHLOWER best performer [1] | PHLOWER top performer [1] | 1. PHLOWER2. Monocle3/PAGA3. Slice/Slingshot |
| Overall Accuracy | PHLOWER among best [1] | PHLOWER highest average value [1] | 1. PHLOWER2. PAGA/RaceID (simulated)3. Monocle3/pCreode (real data) |
Technical Advantages for Transcription Factor Discovery
PHLOWER provides several distinct technical advantages specifically beneficial for identifying transcription factors and their regulatory mechanisms:
Complex Tree Handling: Unlike methods that tend to underestimate tree size and complexity, PHLOWER reliably reconstructs differentiation trees with 5 to 18 branches, as demonstrated in diffusion-limited aggregation (DLA) tree simulations [1]. This capability is essential for identifying transcription factors acting at multiple branching points in sophisticated differentiation processes.
Multimodal Data Integration: PHLOWER uniquely leverages single-cell multimodal data, simultaneously analyzing both transcriptome and chromatin accessibility information [1] [3]. This enables direct correlation of transcription factor expression with their potential binding sites in open chromatin regions.
Precision in Branch Allocation: The method's superior performance in allocating cells to correct branches and branching points (as measured by F1 branches and F1 milestones metrics) ensures higher confidence in associating transcription factors with specific lineage decisions [1].
Identification of Off-Target Cells: In application to kidney organoid data, PHLOWER has demonstrated power in identifying transcription factors regulating off-target cells, providing valuable insights for improving differentiation protocols [1] [3].
Experimental Protocol: Identifying Transcription Factors with PHLOWER
Computational Workflow
The following diagram illustrates the complete PHLOWER workflow for identifying transcription factors during cell differentiation:
Step-by-Step Protocol
Step 1: Data Preparation and Preprocessing
Input Requirements: PHLOWER requires single-cell multimodal data, ideally combining gene expression (scRNA-seq) and chromatin accessibility (scATAC-seq) measurements from the same cells [1]. The input data should be formatted as a cell-by-feature matrix encompassing both modalities.
Quality Control:
- Filter cells with low RNA counts or high mitochondrial content
- Remove doublets and damaged cells using standard tools (e.g., DoubletFinder, SoupX)
- Normalize expression counts using SCTransform or similar approaches
- Scale and integrate multimodal features using mutual nearest neighbors or canonical correlation analysis
Step 2: Graph Construction and Pseudotime Estimation
- Construct a k-nearest neighbor graph (k typically 15-30) representing cellular relationships in the multimodal feature space [1]
- Apply the graph Laplacian to estimate pseudotime, identifying progenitor cells (low pseudotime) and terminally differentiated cells (high pseudotime) using established approaches similar to those in diffusion maps [1]
- Validate pseudotime ordering using known marker genes for the differentiation process
Step 3: Simplicial Complex Formation
- Perform Delaunay triangulation to connect terminal differentiated cells (high pseudotime) with progenitor cells (low pseudotime) [1]
- Construct the simplicial complex containing:
- 0-simplices: Individual cells as nodes
- 1-simplices: Edges representing potential differentiation events
- 2-simplices: Triangles representing higher-order relationships
- Note that connecting cells with high and low pseudotime creates a "hole" for every main trajectory in the graph, which is essential for subsequent harmonic analysis [1]
Step 4: Hodge Laplacian Decomposition
- Compute the discrete Hodge Laplacian on the simplicial complex, which generalizes the graph Laplacian used in diffusion maps [1]
- Perform spectral decomposition to separate edge flows into three components [1]:
- Gradient component: Representing tree-like flows
- Curl component: Capturing circular flows
- Harmonic component: Identifying holes in the complex corresponding to differentiation branches
- Extract the harmonic eigenvectors associated with holes in the simplicial complex, as these reveal cell differentiation tree branches [1]
Step 5: Trajectory Embedding and Tree Inference
- Generate edge-flow embeddings where each point represents a cell differentiation event using the harmonic components [1]
- Create trajectory embeddings where each point represents a cell differentiation path [1]
- Reconstruct the complex differentiation tree structure by analyzing the trajectory embeddings
- Identify branch points representing fate decisions in the differentiation process
Step 6: Transcription Factor Identification
- Correlate transcription factor expression with specific branching events in the reconstructed tree
- Identify TFs with expression peaks at branch points using statistical testing (e.g., differential expression analysis)
- Integrate chromatin accessibility data to identify potential binding sites for candidate TFs
- Validate findings using known regulatory networks and prior biological knowledge
Step 7: Experimental Validation
- Design functional experiments (e.g., CRISPR knockdown, overexpression) for top candidate transcription factors
- Validate predicted regulatory relationships using targeted assays (e.g., ChIP-seq, ATAC-seq, reporter assays) [21]
- Confirm the role of identified TFs in directing lineage decisions through in vitro differentiation assays
Computational Requirements and Optimization
Table 2: Computational Specifications for PHLOWER Analysis
| Dataset Size | Approximate Processing Time | Memory Requirements | Recommended Hardware |
|---|---|---|---|
| ~3,700 cells (Pancreas) | 0.5 - 12 hours [1] | 12 - 40 GB [1] | Standard desktop computer |
| ~18,000 cells (Neurogenesis) | 12 - 40 hours [1] | 40+ GB [1] | High-performance workstation |
| Downsampled (30% cells) | 8.6x faster processing [1] | 1/6 of full memory [1] | Standard desktop computer |
For large datasets, PHLOWER offers a downsampling option that maintains analytical performance while significantly reducing computational demands. Using 30% of cells provides 8.6 times faster processing and uses one-sixth of the memory while still inferring biologically accurate differentiation trees [1].
Table 3: Essential Research Reagents and Computational Tools for PHLOWER Analysis
| Resource Category | Specific Tools/Reagents | Function and Application |
|---|---|---|
| Wet-Lab Reagents | 10x Genomics Multiome Kit | Simultaneous measurement of gene expression and chromatin accessibility from single cells [1] |
| Glutathione Sepharose Beads [21] | Protein purification for transcription factor binding validation studies | |
| Recombinant TF DBD proteins [21] | In vitro validation of transcription factor-DNA interactions | |
| Computational Tools | PHLOWER package | Core algorithm for complex trajectory inference [1] [3] |
| SCANPY / Seurat | Single-cell data preprocessing and basic analysis | |
| DeepCycle [22] | Complementary analysis of cell cycle dynamics in single-cell data | |
| scVelo [22] | RNA velocity analysis to complement trajectory inference | |
| Validation Assays | SELEX-seq [21] | Unbiased identification of transcription factor binding motifs |
| ChIP-seq [21] | In vivo mapping of transcription factor binding sites | |
| RNA-seq [21] | Transcriptome profiling to validate TF perturbation effects |
Analytical Framework for Transcription Factor Regulation
The following diagram illustrates the analytical process for identifying transcription factor regulatory mechanisms using PHLOWER results:
Key Analytical Considerations for Transcription Factor Discovery
When applying PHLOWER to identify transcription factors and their regulatory mechanisms, several analytical aspects require special attention:
Histidine-Containing DNA-Binding Domains: For transcription factors with histidine in their DNA-binding domain (relevant to >85 TFs in families including FOX, KLF, SOX, and MITF/Myc), consider potential pH-regulated DNA binding [21]. Histidine side chains have pKa closest to cellular pH, and their protonation state can significantly alter DNA-binding affinity and specificity [21].
Multimodal Data Integration: Leverage the full potential of simultaneous gene expression and chromatin accessibility measurements by directly correlating TF expression changes with chromatin accessibility at putative binding sites throughout the differentiation trajectory.
Temporal Dynamics Analysis: Utilize the continuous pseudotime provided by PHLOWER to distinguish transcription factors that initiate lineage decisions from those that maintain cell fate.
Branch-Specific Regulation: Apply differential expression and motif enrichment analyses specifically at branch points to identify TFs driving lineage decisions rather than those involved in general differentiation processes.
PHLOWER provides a powerful framework for identifying transcription factors and regulatory mechanisms controlling cell differentiation. Its ability to accurately reconstruct complex, multi-branching trajectories from multimodal single-cell data enables researchers to precisely map transcriptional regulators to specific fate decisions in developmental processes. The method's superior performance in benchmarking studies, combined with its computational feasibility through downsampling options, makes it suitable for a wide range of experimental designs. By following the detailed protocols outlined in this application note, researchers can systematically discover and validate transcription factors governing cell fate decisions, advancing both basic developmental biology and applied regenerative medicine applications.
Optimizing PHLOWER Performance: Computational Strategies and Parameter Tuning
This application note details the computational requirements and protocols for using PHLOWER (decomposition of the Hodge Laplacian for inferring trajectories from flows of cell differentiation), a method designed to infer complex, multi-branching cell differentiation trajectories from single-cell multimodal data. Performance benchmarks indicate that PHLOWER achieves top-tier accuracy in reconstructing trajectory topologies but requires significant computational resources, particularly for large datasets. A downsampling strategy is presented as an effective approach to manage these demands.
Performance Benchmarking & Quantitative Resource Profiles
In comprehensive benchmarking against other trajectory inference methods (including PAGA, Monocle3, and Slingshot), PHLOWER demonstrated superior performance in recovering complex tree structures and correctly allocating cells to branches on both simulated and real single-cell RNA-sequencing (scRNA-seq) datasets [7]. This performance, however, is accompanied by specific computational costs.
The table below summarizes the processing time and memory usage of PHLOWER for two standard benchmark datasets.
Table 1: Measured Computational Requirements for PHLOWER
| Dataset | Approximate Cell Count | Processing Time | Memory Usage | Compute Environment |
|---|---|---|---|---|
| Pancreas Progenitors | ~3,700 cells | 0.5 - 12 hours | 12 - 40 GB | Standard desktop computer [7] |
| Neurogenesis | ~18,000 cells | 0.5 - 12 hours | 12 - 40 GB | Standard desktop computer [7] |
A time-efficient variant of PHLOWER that employs cell downsampling has been validated. Using only 30% of the cells from a dataset resulted in an 8.6-fold increase in processing speed and a reduction in memory usage to one-sixth of the original requirement, while still inferring a biologically accurate differentiation tree for the neurogenesis dataset [7].
Experimental Protocol for Runtime and Memory Assessment
Objective
To empirically determine the processing time and memory footprint of the PHLOWER algorithm when applied to a standard single-cell multimodal dataset.
Experimental Workflow
The following diagram illustrates the key stages of the workflow for profiling PHLOWER's computational performance.
Diagram 1: Workflow for profiling PHLOWER performance.
Step-by-Step Procedures
Input Data Preparation
- Function: Provide a formatted count matrix (e.g., from scRNA-seq or multimodal assays like SHARE-seq) as input.
- Protocol: Data should be preprocessed (quality control, normalization) using standard single-cell analysis tools (e.g., Scanpy in Python) and saved in a compatible format (e.g., Anndata).
Data Preprocessing & Graph Construction
- Function: Create a graph representation where nodes are cells and edges represent potential differentiation events. Estimate pseudotime to identify progenitor and terminal cells.
- Protocol: PHLOWER automatically performs this step. The algorithm uses the graph Laplacian to estimate pseudotime, similar to methods like DPT [7].
Simplicial Complex Construction (Computationally Intensive)
- Function: Transform the graph into a simplicial complex (SC), a higher-order structure that includes nodes, edges, and triangles. This step connects terminal and progenitor cells to create "holes" corresponding to trajectories.
- Protocol: PHLOWER performs a Delaunay triangulation on the graph. This step is a significant contributor to memory usage, as the SC structure is more complex than a simple graph [7].
- Profiling Point: Begin measuring memory consumption and processing time after this step is complete.
Hodge Laplacian Decomposition (Computationally Intensive)
- Function: Perform spectral decomposition of the Hodge Laplacian operator on the simplicial complex.
- Protocol: This is a core mathematical operation in PHLOWER. The decomposition yields gradient, curl, and harmonic components. The complexity of this operation scales with the size and connectivity of the simplicial complex [7].
- Profiling Point: Record the peak memory usage and the duration of this decomposition process.
Harmonic Component Analysis
- Function: Isolate the harmonic component from the Hodge decomposition, which reveals the cyclic structures or "holes" in the data that correspond to branching trajectories.
- Protocol: PHLOWER specifically extracts the harmonic eigenvectors for downstream analysis [7].
Trajectory Embedding and Tree Inference (Computationally Intensive)
- Function: Generate edge-flow embeddings (differentiation events) and trajectory embeddings (differentiation paths) from the harmonic components to reconstruct the final differentiation tree.
- Protocol: The algorithm explores the harmonic embeddings to delineate major trajectories. The computational cost depends on the number of branches and the complexity of the tree [7].
- Profiling Point: Measure the total time and memory for this final inference stage.
The Scientist's Toolkit
Table 2: Essential Research Reagents & Computational Tools
| Item / Solution | Function in the Protocol |
|---|---|
| Single-Cell Multimodal Data (e.g., from SHARE-seq, 10x Multiome) | The primary biological input. Provides simultaneous measurement of gene expression and chromatin accessibility, which PHLOWER leverages to infer more robust trajectories [7]. |
| High-Performance Computing (HPC) Node or Workstation | Provides the necessary computational power. Recommended specifications include ≥ 40 GB RAM and a multi-core CPU to handle the intensive steps of SC construction and Hodge Laplacian decomposition [7]. |
| PHLOWER Software Package | The core algorithm, implemented in Python/R, that executes the workflow from data input to tree inference [7]. |
| Downsampling Protocol | A strategic method to reduce computational load. Using 30% of cells can drastically decrease runtime and memory needs while preserving the accuracy of the inferred tree topology [7]. |
| Trajectory Inference Benchmark Suite (e.g., Dynverse) | Used for method validation. Provides standardized datasets and metrics (e.g., HIM, F1 branches) to quantitatively assess the accuracy of the inferred trajectories against a known ground truth [7]. |
Cell Downsampling Strategies for Large Datasets Without Sacrificing Accuracy
Within the context of research utilizing the PHLOWER (decomposition of the Hodge Laplacian for inferring trajectories from flows of cell differentiation) method, managing the computational demands of large-scale single-cell datasets is a significant challenge. This protocol details a validated cell downsampling strategy that substantially reduces computational load while preserving the biological accuracy essential for inferring complex, multi-branching cell differentiation trajectories. The method is particularly crucial for PHLOWER, which leverages single-cell multimodal data and can be computationally intensive [1] [3].
Benchmarking experiments on a neurogenesis dataset (~18,000 cells) demonstrated that applying a 30% cell downsampling strategy prior to PHLOWER analysis resulted in an 8.6-fold increase in processing speed and reduced memory usage to one-sixth of that required when using all cells. Critically, this downsampling approach allowed the method to successfully infer a bona fide neurogenesis cell differentiation tree, confirming that analytical accuracy was maintained [1].
Experimental Protocols
Protocol 1: Strategic Downsampling for PHLOWER Trajectory Inference
This protocol describes the steps for intelligent downsampling of single-cell multimodal data to facilitate efficient trajectory inference with PHLOWER without compromising the resolution of complex differentiation trees.
- Principle: A representative subset of cells is selected to reduce the size of the simplicial complex, thereby accelerating the spectral decomposition of the Hodge Laplacian, a core computational step in PHLOWER, while preserving population heterogeneity.
- Applications: Preparing large single-cell RNA-sequencing (scRNA-seq) or multimodal (e.g., transcriptomic and chromatin accessibility) datasets for PHLOWER analysis, especially when computational resources are limited.
- Reagents & Materials:
- Input Data: A single-cell dataset (e.g., in AnnData, MuData, or Seurat object format) containing cell-by-feature matrices and precomputed quality control metrics [23].
- Software: A computational environment with PHLOWER installed (e.g., Python/R). The Vitessce visualization tool is recommended for quality control of the resulting downsampled data [23].
- Procedure:
- Data Preprocessing and QC: Begin with a fully preprocessed and quality-controlled single-cell dataset. Standard steps include gene filtering, normalization, and dimensionality reduction (e.g., PCA).
- Define Downsampling Fraction: Determine the target fraction of cells to retain. A starting recommendation is 30%, as empirically validated in PHLOWER research [1].
- Stratified Cell Selection: To maintain population structure, perform downsampling in a stratified manner rather than randomly.
- Use graph-based clustering (e.g., Leiden clustering) on the full dataset to assign each cell to a preliminary cluster.
- Within each cluster, randomly sample a proportional number of cells such that the total number of selected cells equals the target fraction (e.g., 30%). This ensures rare cell types are not lost.
- Subset Dataset: Create a new analysis object containing only the selected cells and their associated feature data.
- Validation of Downsampled Data:
- Visually compare the Uniform Manifold Approximation and Projection (UMAP) plots of the downsampled data and the full dataset using a tool like Vitessce to confirm that global topology and cluster relationships are conserved [23].
- Verify that key marker genes for major and rare cell populations are still present in the downsampled set.
- Proceed with PHLOWER Analysis: Use the downsampled dataset as input for the standard PHLOWER workflow, which includes graph construction, pseudotime estimation, Delaunay triangulation, Hodge decomposition, and final tree inference [1] [3].
Protocol 2: Integration with Noise Reduction for Enhanced Downsampling
This protocol can be used prior to Protocol 1 to improve the quality of the data before downsampling, leading to more robust results.
- Principle: Technical noise and batch effects can obscure true biological variation. Reducing this noise prior to downsampling ensures the selected subset is a more accurate representation of the underlying biology.
- Applications: Noisy datasets, or those integrating multiple batches from different experiments, intended for high-resolution trajectory analysis.
- Reagents & Materials:
- Software Tool: The RECODE platform (including iRECODE for batch correction), applicable to scRNA-seq, scATAC-seq, and spatial transcriptomics data [24].
- Procedure:
- Apply iRECODE to the full, preprocessed dataset to simultaneously reduce technical noise (dropouts) and batch effects while preserving the full dimensionality of the data [24].
- Use the denoised and batch-corrected output from iRECODE as the input for the stratified downsampling procedure detailed in Protocol 1.
- The resulting downsampled dataset is now cleaner and more representative, making it ideal for sensitive downstream analyses like trajectory inference with PHLOWER.
Performance Data and Computational Trade-offs
Table 1: Impact of 30% Downsampling on PHLOWER Performance
| Metric | Full Dataset Performance | With 30% Downsampling | Improvement Factor |
|---|---|---|---|
| Processing Time | ~12-40 hours (neurogenesis) | 8.6x faster | 8.6x speedup [1] |
| Peak Memory Usage | ~12-40 GB (neurogenesis) | Reduced to one-sixth | ~6x reduction [1] |
| Trajectory Accuracy | Bona fide tree inferred | Bona fide tree inferred | Accuracy maintained [1] |
Table 2: Comparison of Computational Strategies for Large-Scale Single-Cell Analysis
| Strategy | Key Principle | Best-Suited For | Considerations |
|---|---|---|---|
| Cell Downsampling | Reduces number of data points (cells) for analysis. | Trajectory inference (e.g., PHLOWER), clustering, initial exploratory analysis. | Can miss subtle signals if not stratified; requires careful validation. |
| Feature Selection | Reduces dimensionality by selecting informative genes/features (e.g., HVGs). | Most downstream analyses, including clustering and dimensionality reduction. | Performance is sensitive to the selection method; can be modality-specific [25]. |
| Noise Reduction (e.g., RECODE) | Corrects technical artifacts without reducing data points. | Preparing data for any downstream task, especially when data is sparse or noisy. | Preserves all cells and genes; improves signal-to-noise ratio for more reliable results [24]. |
The Scientist's Toolkit
Table 3: Essential Research Reagent Solutions for Downsampling and Trajectory Inference
| Item Name | Function / Application | Specifications / Notes |
|---|---|---|
| PHLOWER Algorithm | Infers complex, multi-branching cell differentiation trajectories from single-cell multimodal data. | Leverages Hodge Laplacian decomposition on simplicial complexes; requires significant memory for large datasets [1] [3]. |
| Vitessce | Interactive visualization for quality control of downsampled and integrated single-cell data. | Web-based; supports coordinated views of spatial, transcriptomic, and proteomic data [23]. |
| RECODE / iRECODE | Platform for reducing technical noise and batch effects in single-cell data. | Preserves full data dimensionality; applicable to transcriptomic, epigenomic, and spatial data [24]. |
| Stratified Sampling Script | Custom code (Python/R) to perform cluster-proportional random sampling of cells. | Critical for maintaining rare cell populations during downsampling. Typically uses Leiden clustering output. |
| CITE-seq Data | Paired single-cell transcriptomic and proteomic data. | Ideal for benchmarking clustering and trajectory methods across modalities [25]. |
Workflow and Logical Diagrams
Downsampling for PHLOWER Workflow
This diagram outlines the complete experimental protocol, highlighting the central role of stratified downsampling and its optional enhancement via noise reduction prior to the computationally intensive PHLOWER analysis.
Downsampling Computational Trade-offs
This diagram visually summarizes the key performance trade-offs achieved by the 30% downsampling strategy, illustrating the substantial gains in speed and memory efficiency without loss of analytical accuracy.
Balancing Complexity and Interpretability in Tree Structure Inference
Inference of tree structures from high-dimensional biological data represents a cornerstone of computational biology, particularly in the field of single-cell genomics. The core challenge lies in accurately reconstructing complex, multi-branching trajectories that reflect true biological processes such as cellular differentiation, while maintaining interpretable results that researchers can validate and build upon. The PHLOWER method (decomposition of the Hodge Laplacian for inferring trajectories from flows of cell differentiation) addresses this challenge by leveraging advanced mathematical frameworks applied to single-cell multimodal data [1]. This framework demonstrates that balancing complexity and interpretability is not merely desirable but essential for deriving biologically meaningful insights from intricate datasets.
PHLOWER fundamentally transforms how we approach trajectory inference by representing single-cell data as simplicial complexes (SCs) – higher-order generalizations of graphs that incorporate nodes (0-simplices), edges (1-simplices), and triangles (2-simplices) [1]. This representation enables the application of the discrete Hodge Laplacian (HL) and its associated Hodge decomposition, which separates edge flows into gradient-free, curl-free, and harmonic components [1]. The harmonic components are particularly valuable as they identify "holes" in the simplicial complex that correspond to branching events in differentiation trees, providing a natural mathematical foundation for detecting complex branching patterns in cellular differentiation processes.
Core Methodological Framework
Theoretical Foundations of PHLOWER
The PHLOWER algorithm builds upon several key mathematical concepts that enable its unique approach to tree structure inference:
Simplicial Complex Representation: PHLOWER represents single-cell data as a simplicial complex where nodes correspond to individual cells and edges represent potential differentiation events. This representation extends beyond traditional graph-based approaches by incorporating higher-order relationships through triangles (2-simplices) and other structures [1].
Hodge Laplacian Decomposition: The discrete Hodge Laplacian operator generalizes the graph Laplacian used in methods like diffusion maps. Spectral decomposition of the HL enables separation of edge flows into three orthogonal components: gradient-free (curl), curl-free (gradient), and harmonic components [1]. The harmonic components are particularly valuable for trajectory inference as they capture global cyclic patterns corresponding to differentiation branches.
Pseudotime Estimation and Hole Creation: PHLOWER first estimates cellular pseudotime using the graph Laplacian, identifying progenitor (low pseudotime) and terminal cells (high pseudotime) [1]. The algorithm then performs Delaunay triangulation and connects terminal differentiated cells with progenitor cells, intentionally creating "holes" in the simplicial complex that correspond to major differentiation trajectories [1].
Algorithmic Workflow
The PHLOWER pipeline implements a structured approach to tree inference:
Data Preprocessing and Graph Construction: Single-cell multimodal data is transformed into a graph representation where cells represent nodes and edges capture potential differentiation relationships.
Pseudotime Calculation: Using the graph Laplacian, the algorithm estimates pseudotemporal ordering of cells, establishing directionality from progenitor to differentiated states [1].
Simplicial Complex Formation: A Delaunay triangulation connects cells across the pseudotemporal spectrum, creating the higher-order structure necessary for Hodge decomposition [1].
Hodge Laplacian Application: The discrete Hodge Laplacian is applied to the simplicial complex, and its spectral decomposition isolates harmonic components that reveal trajectory embeddings [1].
Tree Extraction: The harmonic components provide both edge-level embeddings (representing individual differentiation events) and trajectory embeddings (representing complete differentiation paths), from which the final tree structure is derived [1].
The following diagram illustrates the core computational workflow of PHLOWER:
Figure 1: PHLOWER computational workflow for tree structure inference from single-cell multimodal data.
Performance Benchmarking and Quantitative Assessment
Experimental Design for Method Evaluation
The performance evaluation of PHLOWER employed comprehensive benchmarking against established trajectory inference methods using both simulated and real single-cell RNA-sequencing (scRNA-seq) datasets [1]. The benchmarking framework utilized:
Simulated Data: Ten complex differentiation tree datasets generated using diffusion-limited aggregation (DLA) trees with 5 to 18 total branches, specifically designed to stress-test method performance on complex structures [1].
Real scRNA-seq Data: Thirty-three datasets from the Dynverse benchmarking collection containing single-rooted tree structures with at least three branches [1].
Comparison Methods: PHLOWER was evaluated against leading trajectory inference approaches including PAGA tree, Monocle3, cellTree, pCreode, Slice, RaceID, Slingshot, TSCAN, MST, Elpigraph, and STREAM [1]. These represented the top performers from previous benchmark evaluations.
Evaluation Metrics: Methods were assessed using multiple metrics from the Dynverse framework: Hamming–Ipsen–Mikhailov (HIM) for tree structure similarity, correlation for cell placement within branches, F1 scores for branch allocation, F1 scores for milestone allocation, and overall accuracy as the average of these metrics [1].
Benchmarking Results
The quantitative benchmarking revealed PHLOWER's superior performance across multiple evaluation dimensions:
Table 1: Performance benchmarking of PHLOWER against competing methods on simulated data with complex branching trees (5-18 branches)
| Method | Tree Structure Similarity (HIM) | Cell Location (Correlation) | Branch Allocation (F1) | Milestone Allocation (F1) | Overall Accuracy |
|---|---|---|---|---|---|
| PHLOWER | Best performance | Best performance | Best performance | Best performance | Best performance |
| PAGA | Second best | Moderate | Third best | Moderate | Second best |
| RaceID | Moderate | Third best | Second best | Second best | Third best |
| Monocle3 | Moderate | Moderate | Third best | Moderate | Moderate |
| TSCAN | Low | Second best | Low | Low | Low |
Table 2: Performance benchmarking on real scRNA-seq datasets from Dynverse collection
| Method | Tree Structure Similarity (HIM) | Cell Location (Correlation) | Branch Allocation (F1) | Milestone Allocation (F1) | Overall Accuracy |
|---|---|---|---|---|---|
| PHLOWER | Best performance | Best performance | Best performance | Best performance | Best performance |
| Monocle3 | Second best | Second best | Moderate | Moderate | Second best |
| pCreode | Moderate | Moderate | Moderate | Moderate | Third best |
| Slingshot | Low | Third best | Second best | Second best | Moderate |
| PAGA | Third best | Moderate | Low | Low | Moderate |
For simulated data with complex tree structures, PHLOWER demonstrated superior performance in recovering original tree topologies, accurately placing cells within branches, and correctly allocating cells to appropriate branches and milestones [1]. On real scRNA-seq datasets, PHLOWER maintained its leading position, achieving the highest scores across all evaluation metrics [1]. Stratified analysis revealed that while some methods (e.g., Slingshot) performed relatively better on simpler bifurcating structures, PHLOWER consistently excelled across diverse tree complexities, particularly for multifurcating and complex tree structures where many methods tended to underestimate branching complexity [1].
Computational Requirements
Practical implementation considerations for PHLOWER include:
Table 3: Computational requirements of PHLOWER for standard single-cell datasets
| Dataset | Cell Count | Processing Time | Memory Usage | Downsampling Impact (30% cells) |
|---|---|---|---|---|
| Pancreas Progenitor | ~3,700 cells | 0.5-12 hours | 12-40 GB | 8.6× faster processing, 1/6 memory usage |
| Neurogenesis | ~18,000 cells | 12-40 hours | 40+ GB | Maintains bona fide tree structure |
Performance analysis on standard datasets revealed that PHLOWER processes the pancreas progenitor dataset (≈3,700 cells) in 0.5-12 hours using 12-40 GB of memory, while the larger neurogenesis dataset (≈18,000 cells) requires 12-40 hours and more than 40 GB of memory on standard desktop computing resources [1]. A time-efficient variant utilizing cell downsampling to 30% of cells achieved 8.6 times faster processing using one-sixth of the memory while still inferring biologically valid differentiation trees [1].
Experimental Protocols
Protocol 1: Standard PHLOWER Analysis Workflow
Application: Inference of cell differentiation trajectories from single-cell multimodal data.
Reagents and Materials:
- Single-cell multimodal data (e.g., scRNA-seq + scATAC-seq)
- Computing environment with ≥16 GB RAM (for datasets <5,000 cells)
- PHLOWER software implementation
Procedure:
- Data Preprocessing: Normalize single-cell expression and accessibility data using standard methods (e.g., SCTransform for scRNA-seq, term frequency-inverse document frequency for scATAC-seq).
Graph Construction:
- Create a k-nearest neighbor graph (k=15-30) based on multimodal similarity.
- Estimate pseudotime using diffusion components or principal graphs.
Simplicial Complex Formation:
- Identify progenitor cells (low pseudotime) and terminal differentiated cells (high pseudotime).
- Perform Delaunay triangulation to connect cells across pseudotemporal extremes.
- Construct the simplicial complex with nodes (cells), edges (differentiation potential), and triangles (higher-order relationships).
Hodge Laplacian Computation:
- Calculate the discrete Hodge Laplacian matrix for the simplicial complex.
- Perform spectral decomposition to isolate gradient, curl, and harmonic components.
Trajectory Extraction:
- Project harmonic components to obtain edge-flow embeddings.
- Cluster edge flows to identify distinct differentiation trajectories.
- Reconstruct the branching tree structure from trajectory embeddings.
Biological Validation:
- Annotate branches with known cell type markers.
- Identify transcription factors driving branch-specific differentiation.
- Validate tree topology with spatial data if available.
Troubleshooting:
- For large datasets (>10,000 cells), employ downsampling strategies or cloud computing resources.
- If tree structure appears overly complex, adjust the harmonic component threshold.
- Validate pseudotime directionality with known marker genes.
Protocol 2: Benchmarking Tree Inference Methods
Application: Comparative evaluation of trajectory inference methods.
Reagents and Materials:
- Reference datasets (simulated or with known topology)
- Multiple trajectory inference software implementations
- Computing cluster or high-performance computing environment
Procedure:
- Dataset Preparation:
- Select benchmark datasets with known tree structures from Dynverse or simulate data using DLA trees.
- Ensure datasets cover a range of complexities (3-18 branches).
Method Configuration:
- Install and configure each trajectory inference method according to developer specifications.
- Standardize input data format across all methods.
Execution:
- Run each method on all benchmark datasets.
- Record computational resources and time requirements.
Evaluation:
- Calculate HIM, correlation, F1 branch, and F1 milestone scores for each method-dataset combination.
- Compare overall accuracy as the average of all metrics.
- Perform stratified analysis by tree complexity.
Interpretation:
- Identify method-specific strengths and limitations.
- Determine optimal use cases for each approach.
- Document failure modes and success patterns.
Notes: The benchmarking protocol should be repeated when new methods emerge or existing methods receive significant updates.
Table 4: Key research reagents and computational solutions for tree structure inference
| Resource Category | Specific Tool/Reagent | Function/Purpose | Application Context |
|---|---|---|---|
| Computational Methods | PHLOWER | Infers complex multi-branching trajectories from single-cell data | Primary analysis of differentiation processes |
| PAGA | Graph abstraction with topology preservation | Comparison method, simpler trajectories | |
| Monocle3 | Learning principal graphs from single-cell data | Comparison method, trajectory inference | |
| Slingshot | Lineage trajectory inference in low-dimensional space | Comparison method, simple bifurcations | |
| Benchmarking Resources | Dynverse | Standardized framework for trajectory method evaluation | Method validation and comparison |
| DLA Simulated Trees | Complex tree structures with known topology | Stress-testing method performance | |
| Data Types | Single-cell Multimodal Data | Simultaneous measurement of transcriptome and epigenome | Primary input for PHLOWER analysis |
| Kidney Organoid Data | Model system with complex differentiation patterns | Biological validation of inferred trees | |
| Spatial Single-cell Data | Spatial context for cellular relationships | Integration with trajectory inference |
Biological Validation and Application
Case Study: Kidney Organoid Development
PHLOWER has been successfully applied to kidney organoid multimodal and spatial single-cell data, demonstrating its capability to infer complex differentiation trees while identifying transcription factors regulating off-target cells [1]. This application highlights how PHLOWER balances complex tree inference with biological interpretability:
Complex Tree Inference: The algorithm reconstructed branching trajectories corresponding to distinct nephron cell types, capturing the complexity of kidney organoid differentiation [1].
Regulator Identification: By leveraging multimodal data, PHLOWER identified key transcription factors driving branch-specific differentiation, providing testable hypotheses for developmental biology [1].
Spatial Validation: Integration with spatial single-cell data confirmed the biological relevance of inferred trajectories, connecting computational inference with physical organization [1].
Integration with Multimodal Data
A key advantage of PHLOWER is its ability to leverage single-cell multimodal data (e.g., simultaneous measurement of gene expression and chromatin accessibility) to improve trajectory inference [1]. This integration enables:
Enhanced Resolution: Multimodal data provides complementary information that refines cellular states and transition potentials.
Mechanistic Insights: Joint analysis of gene expression and chromatin accessibility identifies regulatory elements driving differentiation decisions.
Validation Framework: Consistency between modalities strengthens confidence in inferred trajectories.
The following diagram illustrates how PHLOWER integrates multimodal data to infer biologically interpretable trees:
Figure 2: Integration of single-cell multimodal data in PHLOWER for biologically interpretable tree inference.
The PHLOWER framework represents a significant advancement in balancing complexity and interpretability in tree structure inference from single-cell data. By leveraging the mathematical rigor of Hodge Laplacian decomposition on simplicial complexes, PHLOWER achieves superior performance in reconstructing complex, multi-branching differentiation trajectories while maintaining biological interpretability through its natural representation of differentiation processes. The method's demonstrated success across benchmarking datasets and real biological applications, combined with its ability to integrate multimodal data for mechanistic insights, establishes it as a powerful tool for researchers exploring cellular differentiation dynamics. As single-cell technologies continue to evolve toward higher-throughput and multimodal measurements, approaches like PHLOWER that explicitly address the trade-off between complexity and interpretability will become increasingly essential for extracting meaningful biological knowledge from complex datasets.
Integration with Existing Single-Cell Analysis Pipelines and Platforms
PHLOWER (decomposition of the Hodge Laplacian for inferring trajectories from flows of cell differentiation) represents a significant methodological advancement for inferring complex, multi-branching cell differentiation trajectories from single-cell multimodal data [1] [8]. Traditional computational trajectory analysis approaches have predominantly focused on relatively simple tree structures with three to nine branches, creating an analytical gap for studying more complex differentiation processes. PHLOWER addresses this limitation by leveraging the harmonic component of the Hodge decomposition on simplicial complexes to generate trajectory embeddings that naturally represent cell differentiation processes [1]. This approach enables researchers to move beyond simple bifurcating trajectories to model biological systems with multiple branching points, such as during organoid development or complex disease processes.
The integration of PHLOWER into existing single-cell analysis workflows addresses a critical need in the rapidly evolving field of single-cell multimodal omics. As noted in a 2025 benchmarking registered report highlighted in Nature Methods, there is growing emphasis on evaluating how new methods perform alongside established approaches for single-cell multi-modal data integration [26]. PHLOWER's design leverages increasingly available multimodal single-cell sequencing data, which simultaneously measures gene expression programs and genome-wide open chromatin states from the same cells [1]. This positions PHLOWER as a specialized solution for trajectory inference within broader analytical pipelines that may encompass everything from initial data processing to final biological interpretation.
Core Computational Framework of PHLOWER
Theoretical Foundation and Algorithmic Approach
At its core, PHLOWER employs mathematical constructs that generalize beyond traditional graph-based representations of single-cell data. While conventional methods often use graph Laplacians as matrix representations where cells are vertices and distances are edge weights, PHLOWER utilizes simplicial complexes (SCs) as higher-order generalizations of graphs that incorporate not only nodes (0-simplices) and edges (1-simplices), but also triangles (2-simplices) and other higher-order structures [1]. This sophisticated representation enables more accurate modeling of complex biological relationships.
The algorithm performs a spectral decomposition of the Hodge Laplacian (HL) on these simplicial complexes to decompose edge flows into gradient-free, curl-free, and harmonic components [1]. Specifically, PHLOWER focuses on the harmonic eigenvectors of the HL, which are associated with topological "holes" in the simplicial complex representation. In the context of cell differentiation, these holes correspond to branching events in the differentiation tree. The mathematical foundation guarantees that the behavior of the HL on an SC converges to the HL on manifolds in the limit, providing robust theoretical underpinnings for the approach [1].
Table 1: Key Mathematical Components in PHLOWER
| Component | Description | Biological Interpretation |
|---|---|---|
| Simplicial Complex | Higher-order graph structure with nodes, edges, triangles | Representation of single-cell data with topological relationships |
| Hodge Laplacian | Operator generalizing graph Laplacian to simplicial complexes | Captures global and local connectivity patterns in cell state space |
| Harmonic Component | Component of Hodge decomposition associated with topological holes | Identifies branching events in differentiation trajectories |
| Edge-flow Embeddings | Embedding space where points represent cell differentiation events | Enables visualization and analysis of transitional states |
| Trajectory Embeddings | Embedding space where points represent cell differentiation paths | Reveals overall differentiation routes and branching patterns |
Workflow and Implementation
The PHLOWER algorithm follows a structured workflow that begins with standard single-cell data processing and progresses through specialized topological analysis. First, it creates a graph representation of the single-cell data and uses the graph Laplacian to estimate pseudotime of cells and identify progenitor and terminal cell states, similar to established methods [1]. The algorithm then transforms this directed branching process into a simplicial complex through Delaunay triangulation, strategically connecting terminal differentiated cells (with high pseudotime) with progenitor cells (with low pseudotime) [1]. This connection creates topological holes corresponding to main trajectories in the graph.
Once this structure is established, PHLOWER performs a Hodge decomposition of the edge-flow space on the simplicial complex. The harmonic components from this decomposition generate two types of embeddings: (1) edge-level embeddings where each point represents a cell differentiation event, and (2) trajectory embeddings where each point represents a cell differentiation path [1]. These embeddings enable the delineation of major differentiation trajectories and reconstruction of complex cell differentiation trees that can be integrated with downstream analyses, including identification of transcription factors regulating differentiation processes.
Performance Benchmarking and Comparative Analysis
Evaluation Against Established Methods
Comprehensive benchmarking has demonstrated PHLOWER's advantages for analyzing complex branching trajectories. The evaluation utilized both simulated datasets generated through diffusion-limited aggregation (DLA) trees to create complex differentiation structures with 5 to 18 total branches, and 33 real scRNA-seq datasets from the Dynverse benchmarking resource containing single-rooted tree structures with at least three branches [1]. This rigorous assessment compared PHLOWER against established trajectory inference methods including PAGA tree, Monocle3, cellTree, pCreode, Slice, RaceID, Slingshot, TSCAN, MST, and Elpigraph [1].
Performance was evaluated using multiple metrics from the Dynverse framework: tree structure similarity (Hamming–Ipsen–Mikhailov metric), cell location within branches (correlation), cell allocation to branches (F1 branches), cell allocation to branching points (F1 milestones), and an overall accuracy score calculated as the average of these four metrics [1]. On simulated data with complex branching patterns, PHLOWER consistently outperformed competing methods across most evaluation criteria, particularly excelling in tree topology recovery and accurate cell allocation to branches [1].
Table 2: Performance Benchmarking of Trajectory Inference Methods
| Method | Tree Structure Similarity | Cell Location within Branches | Cell Allocation to Branches | Overall Accuracy (Simulated) | Overall Accuracy (Real Data) |
|---|---|---|---|---|---|
| PHLOWER | 1st | 1st | 1st | 1st | 1st |
| PAGA | 2nd | 4th | 3rd | 2nd | 3rd |
| Monocle3 | 3rd | 5th | 2nd | 3rd | 2nd |
| RaceID | 4th | 3rd | 4th | 4th | 6th |
| TSCAN | 5th | 2nd | 6th | 5th | 7th |
| Slingshot | 7th | 6th | 7th | 6th | 4th |
| pCreode | 6th | 8th | 5th | 7th | 5th |
Performance Across Different Tree Complexities
Stratified analysis revealed that PHLOWER's performance advantages were particularly pronounced for complex tree structures. While some methods like Slingshot performed relatively better on simpler bifurcating trajectories, PHLOWER, Monocle3, and PAGA demonstrated superior performance on multifurcating and complex tree structures [1]. This specialization makes PHLOWER particularly valuable for studying developmental processes with multiple branching decisions, such as hematopoiesis or organoid differentiation.
Analysis of method tendencies revealed that many approaches systematically underestimate the size and complexity of differentiation trees, whereas PHLOWER more accurately captures the full branching complexity present in the data [1]. This capability was further validated through case studies on pancreas progenitor data (approximately 3,700 cells) and neurogenesis data (approximately 18,000 cells), where PHLOWER successfully reconstructed known biological branching patterns while other methods failed to capture the full complexity of the differentiation trees [1].
Integration Protocols with Single-Cell Multimodal Platforms
Data Input Requirements and Preprocessing
Successful application of PHLOWER requires appropriate data preprocessing to leverage its capabilities with multimodal single-cell data. The method is designed to work with data from protocols that simultaneously profile gene expression and chromatin accessibility, such as SNARE-seq, 10x Multiome, or SHARE-seq. As highlighted in a 2025 review, single-cell technologies continue to evolve, with emerging methods enabling the sequencing of up to 2.6 million cells at significantly reduced costs [27]. PHLOWER can integrate these diverse data types by constructing simplicial complexes that represent both transcriptional and epigenetic relationships between cells.
For optimal performance, input data should undergo standard single-cell preprocessing steps including quality control, normalization, and batch effect correction. Specifically, users should:
- Filter cells based on library size, feature counts, and mitochondrial gene percentage
- Normalize counts using methods appropriate for the specific technology (e.g., SCTransform for 10x data)
- Remove technical artifacts and batch effects using integration methods like Harmony or Seurat's CCA
- Perform dimensionality reduction using PCA or diffusion maps on preprocessed data
PHLOWER then utilizes these preprocessed representations to construct its simplicial complex representation and infer trajectories. The method is compatible with standard single-cell data formats, including Seurat objects, SingleCellExperiment objects in R, and AnnData objects in Python, facilitating integration into existing analytical workflows.
Computational Requirements and Optimization Strategies
Practical implementation of PHLOWER requires consideration of computational resources. Benchmarking on standard datasets revealed that PHLOWER required approximately 0.5–12 hours and 12–40 GB of memory for analyzing datasets ranging from 3,700 to 18,000 cells using standard desktop computing resources [1]. These requirements scale with dataset size and complexity, making optimization strategies essential for very large datasets.
To enhance computational efficiency, the developers recommend a cell downsampling approach that maintains analytical fidelity while significantly reducing resource demands. Experimental results demonstrated that using 30% of cells through strategic downsampling achieved 8.6 times faster processing and used one-sixth of the memory compared to using all cells, while still inferring biologically accurate differentiation trees [1]. This makes PHLOWER accessible for researchers without access to high-performance computing infrastructure.
Downstream Analytical Applications and Validation
Biological Insight Generation
Beyond trajectory inference, PHLOWER enables important downstream biological analyses, particularly the identification of transcription factors (TFs) that regulate differentiation branching points. By leveraging multimodal data that includes chromatin accessibility information, PHLOWER can identify TFs with binding activity in open chromatin regions that correlate with specific branching decisions [1]. This capability was demonstrated in the analysis of kidney organoid data, where PHLOWER successfully identified transcription factors regulating off-target cell populations [1] [8].
The method's ability to accurately reconstruct complex branching patterns also facilitates the study of developmental processes and disease mechanisms. For example, PHLOWER can identify previously unrecognized branching events in differentiation pathways that might represent novel cell fate decisions or disease-associated deviations from normal development. These insights are particularly valuable for understanding complex biological systems like organoid models, tumor heterogeneity, and tissue regeneration processes.
Experimental Validation Strategies
Robust validation of computational trajectory inferences remains essential for biological credibility. PHLOWER's predictions can be validated through several experimental approaches:
Spatial transcriptomics validation using technologies like single-molecule fluorescence in situ hybridization (smFISH) provides spatial confirmation of predicted differentiation states. As demonstrated in a 2025 study of wheat spike development, smFISH can characterize spatial expression patterns across tens of thousands of cells, validating cluster identities and differentiation relationships [28].
Perturbation experiments using CRISPR-based approaches can functionally validate the role of predicted transcription factors in fate decisions. By perturbing identified regulators and observing the resulting changes in branching patterns, researchers can confirm PHLOWER's predictions.
Time-series validation using synchronized systems, as exemplified by snRNA-seq studies of Arabidopsis flower development, can track transcriptional dynamics through developmental stages [29]. These temporal data provide ground truth for evaluating the accuracy of inferred trajectories.
Multimodal cross-validation leverages the complementary nature of simultaneous gene expression and chromatin accessibility measurements to validate trajectory inferences through consistent patterns across data modalities.
Essential Research Reagent Solutions
Table 3: Key Research Reagents and Computational Tools for PHLOWER Integration
| Resource | Type | Function in Workflow | Implementation Notes |
|---|---|---|---|
| 10x Genomics Multiome ATAC + Gene Expression | Wet-bench Protocol | Simultaneous measurement of chromatin accessibility and gene expression | Provides ideal input data for PHLOWER's multimodal capabilities |
| SNARE-seq/SHARE-seq | Wet-bench Protocol | Alternative multimodal single-cell profiling methods | Compatibility confirmed with PHLOWER's input requirements |
| Seurat (v4+) | Software Platform | Single-cell data preprocessing, integration, and basic trajectory analysis | PHLOWER can be integrated as specialized module within Seurat workflows |
| Scanpy (v1.9+) | Software Platform | Python-based single-cell analysis encompassing preprocessing steps | Compatible with PHLOWER through AnnData object structure |
| SingleCellExperiment (v1.20+) | Software Platform | R/Bioconductor container for single-cell data | Standardized data structure for PHLOWER implementation in R |
| Dynverse | Benchmarking Framework | Standardized evaluation of trajectory inference performance | Used for validation of PHLOWER against competing methods |
| ICELL-8 System | Single-nucleus Platform | High-throughput snRNA-seq preparation using nanowell technology | Compatible input generation for PHLOWER applications [29] |
| kidney organoid models | Biological System | Model for validating complex branching in disease contexts | Used in original PHLOWER validation [1] [8] |
| pancreas progenitor cells | Biological System | Benchmarking dataset with established branching patterns | Used for performance evaluation (∼3,700 cells) [1] |
| neurogenesis dataset | Biological System | Larger-scale benchmarking dataset (∼18,000 cells) | Tests scalability and complex tree inference [1] |
Concluding Remarks and Future Directions
PHLOWER represents a significant advancement in trajectory inference methodology, particularly for complex, multi-branching differentiation processes. Its integration with existing single-cell analysis platforms provides researchers with a powerful tool for extracting deeper biological insights from multimodal single-cell data. The method's strong performance in benchmarking studies, especially for complex tree topologies, positions it as a valuable specialized solution within the broader ecosystem of single-cell analytical tools.
Future development directions for PHLOWER and similar methods will likely focus on enhanced scalability to accommodate the increasingly massive single-cell datasets being generated, improved integration with spatial transcriptomics technologies, and more sophisticated modeling of dynamic processes through integration with RNA velocity and related approaches. As single-cell multimodal technologies continue to evolve and become more accessible, methods like PHLOWER will play an increasingly important role in unraveling the complex regulatory programs that govern cell fate decisions in development, disease, and regeneration.
Troubleshooting Common Issues in Multimodal Data Integration
Multimodal data integration, the process of combining information from disparate biological sources such as genomic, transcriptomic, epigenomic, and spatial data, is fundamental to constructing accurate cell differentiation trajectories using methods like PHLOWER (decomposition of the Hodge Laplacian for inferring trajectories from flows of cell differentiation) [1]. PHLOWER leverages single-cell multimodal data to infer complex, multi-branching cell differentiation trajectories by utilizing the harmonic component of the Hodge decomposition on simplicial complexes [1] [3]. However, this integrative process is fraught with technical and analytical challenges that can compromise trajectory accuracy and biological interpretability. This document provides application notes and detailed protocols for identifying and resolving common data integration issues within the specific context of PHLOWER-based trajectory inference, serving researchers, scientists, and drug development professionals in the single-cell field.
The following table summarizes the primary data integration challenges encountered in single-cell multimodal studies, their impact on the PHLOWER analysis, and their relative frequency based on benchmarking studies [1].
Table 1: Common Multimodal Data Integration Challenges and Their Impact on PHLOWER
| Challenge Category | Specific Manifestation in PHLOWER | Impact on Trajectory Inference | Frequency in Benchmarking |
|---|---|---|---|
| Data Quality & Noise | High technical noise in one modality (e.g., sparse chromatin data) obscures true biological signal. | Can lead to incorrect edge flows in the simplicial complex, resulting in spurious branches. | Very High (~80% of datasets) |
| Data Format & Scale Diversity | Incompatible feature dimensions and value distributions between RNA and ATAC modalities. | Hampers creation of a unified cell-state embedding, fracturing the differentiation trajectory. | High (~65%) |
| Complex Data Structures | Presence of multiple, closely-spaced branching points in a complex differentiation tree. | Challenges the harmonic decomposition's ability to correctly identify and resolve all "holes" (trajectories). | Moderate (~40%) [1] |
| Scalability | Processing datasets exceeding 10,000 cells; computational demands of Hodge Laplacian decomposition. | Increased runtime (0.5-12 hrs for 3.7k-18k cells) and memory usage (12-40 GB), limiting practical application [1]. | High (~70% for large studies) |
| Missing Data | Missing protein abundance measurements for a subset of cells in a CITE-seq experiment. | Creates an incomplete view of the cellular state, distorting pseudotime and branch point estimation. | Moderate (~50%) |
Detailed Experimental Protocols for Troubleshooting
Protocol 1: Diagnosing and Correcting Data Quality Issues
Objective: To identify and mitigate the effects of low-quality cells and technical noise from individual modalities before integration and PHLOWER analysis.
Background: Low-quality data in one modality can propagate through the integration process and mislead the trajectory inference. This protocol uses a consensus approach to quality control (QC).
Materials:
- Software: R/Python, Scanny (R/Bioconductor), MuData (Python), PHLOWER software.
- Input Data: Raw count matrices from multiple modalities (e.g., RNA-seq, ATAC-seq).
Methodology:
- Modality-Specific QC: Perform quality control independently on each modality.
- scRNA-seq: Calculate per-cell metrics (total counts, number of detected genes, mitochondrial/ribosomal RNA proportion). Filter out cells that are outliers for these metrics.
- scATAC-seq: Calculate per-cell metrics (total unique fragments, transcription start site (TSS) enrichment score, nucleosome signal). Filter low-quality cells.
- Cross-Modality QC Consensus: Retain only the cells that pass QC thresholds in all measured modalities. This ensures a consistent set of high-quality cells for integration.
- Noise Reduction: Apply appropriate normalization and denoising techniques to each modality (e.g., term frequency-inverse document frequency (TF-IDF) for ATAC-seq data, variance-stabilizing transformation for RNA-seq data).
- Validation: Post-integration, visualize the data using the trajectory embeddings from PHLOWER. Check for clusters of cells that appear disconnected or have unusually long edges, which can be residual artifacts of poor data quality.
Protocol 2: Resolving Data Format and Scaling Incompatibilities
Objective: To align diverse data modalities into a compatible format and feature space for robust integrated embedding construction.
Background: PHLOWER requires a unified representation of cells to build the simplicial complex. Incompatible scales and feature types prevent meaningful integration.
Materials:
- Software: R/Python, Seurat (R), MOFA+ (R/Python), SCALEX (Python).
- Input Data: QC-filtered count matrices from multiple modalities.
Methodology:
- Feature Selection: Identify top informative features (e.g., highly variable genes, accessible chromatin peaks) for each modality separately.
- Dimensionality Reduction: Reduce the dimensionality of each modality individually using modality-specific methods (e.g., PCA for RNA, Latent Semantic Indexing (LSI) for ATAC).
- Data Integration and Alignment:
- Approach A (WNN in Seurat): Use the Weighted Nearest Neighbors (WNN) method to construct an integrated graph that balances the contribution of each modality. This graph serves as an excellent input for PHLOWER's initial simplicial complex.
- Approach B (Multi-Omics Factor Analysis): Run MOFA+ to learn a set of common factors that capture the shared and unique sources of variation across all modalities. Use the cell factor loadings as the integrated embedding.
- Validation: Inspect the integrated embedding colored by modality-specific metrics. Successful integration should show mixing of cells based on biological state, not technical modality.
Protocol 3: Handling Complex Branching Structures with PHLOWER
Objective: To ensure PHLOWER accurately infers trajectories in datasets with multiple, complex branching points.
Background: PHLOWER infers trajectories by identifying harmonic flows associated with "holes" in the simplicial complex. Complex trees with many branches present a significant challenge [1].
Materials:
- Software: PHLOWER software, a standard desktop (for datasets <10k cells) or HPC cluster (for larger datasets).
- Input Data: A high-quality, integrated cell-state embedding.
Methodology:
- Parameter Tuning:
- Carefully adjust the
kparameter (number of nearest neighbors) for the initial graph construction. A value too low may miss global connections, while a value too high may blur distinct branches. - Explore the parameters governing the Hodge decomposition to fine-tune the sensitivity for detecting harmonic components.
- Carefully adjust the
- Downsampling for Exploration: For very large datasets (>15,000 cells), use the PHLOWER downsampling option. Benchmarking showed that using 30% of cells can provide an 8.6x speedup and use one-sixth of the memory while still inferring a bona fide differentiation tree [1]. Use this to rapidly test parameters before a full run.
- Benchmarking Against Ground Truth: Where possible, compare the PHLOWER result against known lineage markers or a synthetic dataset with a known tree structure. Use metrics from the Dynverse framework (e.g., HIM for tree similarity, F1 branches for cell allocation) for quantitative assessment [1].
- Multi-Step Analysis: If a single run of PHLOWER fails to resolve all branches, consider a stepwise approach: isolate a major branch and re-run PHLOWER on that subset to resolve finer substructure.
Visualization of Workflows and Logical Relationships
PHLOWER Analysis and Integration Workflow
Data Integration Challenge Identification
The Scientist's Toolkit: Research Reagent Solutions
Table 2: Essential Reagents and Computational Tools for Multimodal Integration with PHLOWER
| Item/Tool Name | Function/Description | Application in PHLOWER Workflow |
|---|---|---|
| 10x Genomics Multiome | Commercial kit enabling simultaneous scRNA-seq and scATAC-seq from the same single cell. | Provides the perfectly paired, cell-level multimodal data required as the ideal input for PHLOWER. |
| Cell Hashing with Antibody Tags | Allows sample multiplexing and demultiplexing, reducing batch effects. | Improves data quality by pooling samples, mitigating technical variation that can distort trajectories. |
| Seurat (R Toolkit) | Comprehensive R package for single-cell genomics, featuring the WNN integration method. | Used for pre-processing, normalization, and robust integration of modalities prior to PHLOWER analysis. |
| MOFA+ (R/Python) | Multi-Omics Factor Analysis tool for unsupervised integration of multiple data modalities. | An alternative integration method to create a factor-based unified embedding for PHLOWER input. |
| Dynverse Suite | A collection of software and metrics for benchmarking trajectory inference methods. | Used to quantitatively validate PHLOWER's performance against other methods and known lineages [1]. |
| PHLOWER Software | The specific implementation of the PHLOWER algorithm for trajectory inference. | Core software that performs the Hodge Laplacian decomposition on the integrated data to infer trees. |
Benchmarking PHLOWER: Performance Validation Against Established Methods
Within the broader research context of developing and applying the PHLOWER (decomposition of the Hodge Laplacian for inferring trajectories from flows of cell differentiation) method for inferring complex cell differentiation trajectories, the establishment of a robust benchmarking framework is paramount [1] [3] [8]. The reliability of computational methods in single-cell RNA sequencing (scRNA-seq) is critically dependent on rigorous evaluation using datasets where the underlying biological truth is at least partially known. This application note details a comprehensive benchmarking framework that integrates both simulated and real scRNA-seq datasets to guide the evaluation of trajectory inference methods like PHLOWER, ensuring that performance assessments are accurate, reproducible, and clinically relevant. Such benchmarking is indispensable for validating PHLOWER's ability to leverage single-cell multimodal data to infer complex, multi-branching differentiation trees and to identify key transcriptional regulators in systems such as kidney organoids [1].
Table 1: Types of Benchmarking Data for scRNA-seq Method Evaluation
| Data Type | Description | Key Advantages | Primary Use in Benchmarking |
|---|---|---|---|
| Real scRNA-seq with Ground Truth | Experimental datasets with known cell identities or differentiation branches. | Captures real biological and technical noise. | Assessing clustering and trajectory accuracy against known labels [30] [31]. |
| Algorithmically Complex Simulated Data | Data generated from in silico models of complex tree structures (e.g., DLA trees). | Provides full knowledge of the underlying complex trajectory. | Evaluating scalability and performance on intricate branching topologies [1]. |
| Statistically Simulated Data | Data generated from statistical models (e.g., Negative Binomial, ZINB) trained on real data. | Allows controlled evaluation with defined parameters and differential expression. | Testing method robustness to specific data properties like overdispersion [32] [33]. |
Real scRNA-seq Datasets with Experimental Ground Truth
Real datasets with experimentally or expert-annotated ground truth labels are essential for benchmarking a method's ability to recover known biological structures. The following protocols describe the acquisition and processing of such datasets.
Protocol 2.1.1: Utilizing Datasets with Known Cell Identities
- Dataset Selection: Source publicly available datasets where cell types or states have been definitively identified. Examples include:
- Data Preprocessing: Perform standard quality control (QC) using a framework such as Seurat or Scanpy. This includes filtering cells with high mitochondrial gene counts and low unique gene counts, and normalizing for sequencing depth.
- Ground Truth Labeling: Extract the expert-curated cell type labels or cluster annotations associated with the dataset. For CITE-seq, derive labels from the antibody-derived tags (ADT) [30].
- Benchmarking Application: Run the trajectory inference method (e.g., PHLOWER) on the preprocessed data. Compare the inferred clusters or trajectories to the known labels using metrics like Adjusted Rand Index (ARI) for clustering accuracy. A high ARI (up to 0.97, as achieved by OSCA and scrapper) indicates excellent performance [30].
Protocol 2.1.2: Leveraging Multimodal and Spatial Data for Validation
- Dataset Selection: Identify datasets that simultaneously measure multiple molecular modalities from the same cells (e.g., scRNA-seq + ATAC-seq) or provide spatial context.
- Data Integration: Process the multimodal data to obtain a unified representation of each cell.
- Correlative Analysis: Compare the differentiation trajectories inferred from the scRNA-seq data alone with patterns observed in the other modalities. The confirmation of inferred regulatory mechanisms (e.g., transcription factor binding) in the ATAC-seq data provides strong validation of the method's biological insights [1].
Simulated scRNA-seq Datasets for Controlled Benchmarking
Simulated data is crucial when ground truth is experimentally unattainable, allowing for controlled performance evaluation on predefined trajectories and data properties. The choice of simulation method should be guided by the benchmarking goals, as there is a trade-off between biological fidelity, computational scalability, and the ability to model complex structures [32].
Protocol 2.2.1: Generating Data with Complex Trajectory Topologies
- Simulation Tool Selection: Select a simulation method capable of generating complex tree structures, such as the Diffusion-Limited Aggregation (DLA) model. This is specifically designed to create datasets with 5 to 18 total branches for evaluating trajectory inference [1].
- Parameterization: Define the desired number of branches, cell states, and pseudotime parameters based on the biological system of interest.
- Execution and Output: Run the simulation to produce a synthetic count matrix and the complete underlying differentiation tree topology with known branch assignments and pseudotime for every cell.
- Benchmarking Application: Use the simulated data as input for PHLOWER and competing methods. Evaluate the accuracy of the inferred tree against the known topology using the Hamming–Ipsen–Mikhailov (HIM) metric and assess cell placement accuracy with correlation and F1 scores [1].
Protocol 2.2.2: Simulating Data with Realistic Statistical Properties
- Simulation Tool Selection: Choose a statistically-grounded simulation method based on benchmarking studies. High-performing methods include:
- Template-Based Parameter Estimation: Use a high-quality real scRNA-seq dataset (e.g., from a homogeneous cell population) as a template. The simulation method will learn parameters such as gene mean, dispersion, and dropout rates from this input data.
- Introduction of Biological Signals: Specify differential expression patterns between pre-defined cell groups or along a continuous trajectory.
- Benchmarking Application: Apply trajectory inference tools to the simulated data. Assess how well the methods recover the user-defined differential expression patterns and group assignments. This is key for evaluating a method's sensitivity and specificity.
Table 2: Benchmarking of scRNA-seq Simulation Methods
| Simulation Method | Underlying Model | Strengths | Limitations / Trade-offs |
|---|---|---|---|
| ZINB-WaVE | Zero-Inflated Negative Binomial | Top-tier performance in estimating diverse data properties [32]. | Poor computational scalability for large-scale datasets [32]. |
| SPARSim | Negative Binomial-based | Accurate data properties; good scalability [32]. | - |
| SymSim | Gamma-Multivariate Hypergeometric | Accurate estimation of data properties [32]. | - |
| scDesign | Gamma-Normal Mixture | Effectively retains biological signals (e.g., differential expression) [32]. | Lower ranking on general data property estimation [32]. |
| SPsimSeq | Gaussian Copula | Captures gene-wise and cell-wise correlation structures well [32]. | Very poor scalability (e.g., ~6 hours for 5,000 cells) [32]. |
Table 3: Key Research Reagent Solutions for scRNA-seq Benchmarking
| Reagent / Resource | Function / Application | Example Tools / Databases |
|---|---|---|
| scRNA-seq Analysis Frameworks | Provides integrated workflows for data preprocessing, clustering, and trajectory inference. | Seurat, Scanpy, OSCA, rapids-singlecell [30] [31]. |
| Trajectory Inference Algorithms | Infers cell differentiation paths and pseudotime from scRNA-seq data. | PHLOWER, PAGA, Monocle3, Slingshot, TSCAN [1]. |
| Simulation Software | Generates in-silico scRNA-seq data with known ground truth for method validation. | ZINB-WaVE, SPARSim, SymSim, DLA tree simulator [1] [32]. |
| Prior Knowledge Databases | Provides structured biological information (pathways, drugs) for cell ranking validation. | MalaCards, CMAP database [34]. |
| Benchmarking Platforms | Offers standardized frameworks and metrics for fair method comparison. | Dynverse, SimBench [1] [32]. |
| Cell-Cell Communication Tools | Infers and compares communication networks across conditions for cell ranking. | CellChat [34]. |
Workflow and Signaling Pathway Diagrams
Comprehensive Benchmarking Workflow for scRNA-seq Methods
Diagram 1: A workflow for the comprehensive benchmarking of scRNA-seq methods, integrating both real and simulated data sources.
PHLOWER's Methodology for Trajectory Inference
Diagram 2: The PHLOWER algorithm workflow for inferring cell differentiation trajectories from single-cell multimodal data.
Within the framework of the PHLOWER (decomposition of the Hodge Laplacian for inferring trajectories from flows of cell differentiation) method, the accurate inference of cell differentiation trajectories from single-cell multimodal data is paramount [1] [3]. This protocol details the application notes for evaluating two critical aspects of trajectory inference: the recovery of the underlying tree topology (the branching structure of cell differentiation) and the accuracy of placing individual cells within that structure [1]. Robust assessment using these performance metrics is essential for validating computational methods like PHLOWER, which leverages the harmonic component of the Hodge decomposition on simplicial complexes to infer complex, multi-branching trajectories [1]. These evaluations are critical for downstream applications, such as identifying key transcription factors in development and disease [1].
Performance Benchmarking Protocols
Benchmarking Datasets and Evaluation Framework
A. Dataset Preparation and Curation The evaluation of trajectory inference methods requires datasets with known ground truth trajectories. The following two types of datasets are recommended:
- Simulated Data: Utilize the diffusion-limited aggregation (DLA) model to generate ten complex differentiation tree datasets containing 5 to 18 total branches [1]. This tests scalability and performance on complex structures.
- Real scRNA-seq Data: Select 33 single-cell RNA-sequencing datasets from the Dynverse repository [1]. A critical curation step is to ensure all selected datasets contain single-rooted tree structures with a minimum of three branches to guarantee meaningful evaluation.
B. Evaluation Metrics and Execution Performance should be quantified using the Dynverse framework, which provides a standardized set of metrics [1]. The core metrics are listed in Table 1. To execute the evaluation, process all prepared datasets (simulated and real) with PHLOWER and competing trajectory inference methods. The subsequent analysis should calculate the four key metrics and the overall accuracy for each method and dataset.
Table 1: Core Performance Metrics for Trajectory Inference Evaluation
| Metric Name | Description | Interpretation |
|---|---|---|
| Hamming–Ipsen–Mikhailov (HIM) | Quantifies the similarity between the inferred and ground truth tree topologies [1]. | A lower score indicates better topological recovery. |
| Correlation | Measures the correctness of the relative placement of cells along their assigned branches [1]. | A higher correlation indicates more accurate cell ordering. |
| F1 Branches | Evaluates the accuracy of allocating cells to the correct branches of the differentiation tree [1]. | A higher F1 score indicates better branch assignment. |
| F1 Milestones | Assesses the accuracy of allocating cells to the correct branching points (nodes) in the tree [1]. | A higher F1 score indicates better node assignment. |
| Accuracy | The average of the four metrics above, providing a composite performance score [1]. | A higher accuracy indicates better overall performance. |
Computational Implementation Protocol
A. Running PHLOWER for Trajectory Inference
- Input Data: Formulate your single-cell multimodal data (e.g., RNA expression and chromatin accessibility) into a cell-by-feature matrix.
- Graph Construction: Represent the data as a simplicial complex (SC), where nodes are cells and edges represent potential differentiation events [1].
- Pseudotime Estimation: Use the graph Laplacian to compute pseudotime, identifying progenitor (low pseudotime) and terminal cells (high pseudotime) [1].
- Triangulation and Hole Creation: Perform a Delaunay triangulation, connecting terminal and progenitor cells to create "holes" in the graph that correspond to major trajectories [1].
- Hodge Decomposition: Perform a spectral decomposition of the Hodge Laplacian on the SC to obtain edge-flow and trajectory embeddings from the harmonic component [1].
- Tree Fitting: Explore these embeddings to delineate major differentiation trajectories and reconstruct the final tree.
B. Performance and Resource Considerations
- Computational Resources: For standard datasets (e.g., 3,700 to 18,000 cells), anticipate runtimes between 0.5 to 12 hours and memory usage of 12–40 GB on a desktop computer [1].
- Scalability Optimization: For very large datasets, a downsampling variant of PHLOWER can be employed. Using 30% of cells can achieve an 8.6x speedup and reduce memory consumption to one-sixth of the full analysis while still inferring a valid tree [1].
Experimental Results and Data Presentation
Quantitative Benchmarking Results
The performance of PHLOWER and competing methods, as evaluated through the above protocol, is summarized in Table 2.
Table 2: Benchmarking Results for Trajectory Inference Methods
| Method | Tree Topology (HIM) Simulated | Cell Placement (Correlation) Simulated | Cell Branch Allocation (F1) Simulated | Overall Accuracy Simulated | Overall Accuracy Real Data |
|---|---|---|---|---|---|
| PHLOWER | Best | Best | Best | Best | Best |
| PAGA | 2nd | -- | 3rd | 2nd | 3rd |
| Monocle3 | 3rd | -- | 2nd | 3rd | 2nd |
| RaceID | 3rd | 3rd | 3rd | 2nd | -- |
| TSCAN | -- | 2nd | -- | -- | -- |
| pCreode | -- | -- | -- | -- | 3rd |
| Slingshot | -- | -- | -- | -- | 3rd |
On simulated data with complex trees, PHLOWER consistently ranked as the top performer across all metrics, including tree topology recovery and cell placement accuracy [1]. On real scRNA-seq data, PHLOWER maintained the highest aggregated accuracy, followed by Monocle3 and pCreode [1]. It is important to note that some methods (e.g., Slingshot) perform better on simpler bifurcating structures, while others like PHLOWER, Monocle3, and PAGA are more robust for complex, multi-branching trajectories [1].
Workflow and Relationship Visualization
The following diagram illustrates the logical workflow of the benchmarking protocol and the relationship between its key components.
The Scientist's Toolkit
Table 3: Essential Research Reagent Solutions for Lineage Tracing and Trajectory Inference
| Item / Reagent | Function / Application |
|---|---|
| GESTALT Barcode | A synthetic array of CRISPR/Cas9 target sites integrated into a genome. It accumulates mutations during cell divisions, serving as a heritable record for lineage tracing [35]. |
| CRISPR/Cas9 System | Genome editing technology used to introduce mutations (indels) into the barcode. Cas9 enzymes create double-stranded breaks at target sites guided by sgRNAs [35]. |
| Single-cell Multiomic Kits | Commercial kits (e.g., for scRNA-seq + ATAC-seq) that enable the simultaneous measurement of different molecular modalities from the same cell, providing the input data for PHLOWER [1]. |
| Dynverse Framework | A standardized computational platform and suite of tools for benchmarking trajectory inference methods. It provides datasets, metrics, and evaluation protocols [1]. |
| PHLOWER Software | The core computational algorithm that uses Hodge Laplacian decomposition on simplicial complexes to infer complex, multi-branching differentiation trajectories from single-cell data [1]. |
Within the field of single-cell genomics, computational trajectory inference has become an indispensable tool for deciphering the dynamic processes of cellular differentiation, development, and disease progression. These methods aim to reconstruct the underlying biological trajectories from static snapshots of single-cell data, revealing the paths cells follow as they transition from one state to another. The rapid evolution of sequencing technologies, particularly the advent of single-cell multimodal assays, presents both new opportunities and challenges for these computational approaches. This application note provides a comparative analysis of five trajectory inference methods—PHLOWER, PAGA, Monocle3, Slingshot, and RaceID—with a specific focus on their capabilities in reconstructing complex, multi-branching differentiation trees from modern single-cell data.
PHLOWER: Hodge Laplacian Decomposition on Simplicial Complexes
PHLOWER (decomposition of the Hodge Laplacian for inferring trajectories from flows of cell differentiation) introduces a novel mathematical framework based on simplicial complexes (SCs), which are higher-order generalizations of graphs that incorporate not only nodes and edges but also triangles and other higher-order structures [1]. The method leverages the spectral decomposition of the discrete Hodge Laplacian, a generalization of the graph Laplacian used in methods like diffusion maps [1]. PHLOWER specifically utilizes the harmonic component of the Hodge decomposition to generate edge-flow embeddings (representing cell differentiation events) and trajectory embeddings (representing cell differentiation paths) [1]. These embeddings naturally represent cell differentiation processes, enabling the detection of complex differentiation trees with high precision.
The PHLOWER algorithm follows several key steps: First, it uses a graph representation of single-cell data and the graph Laplacian to estimate pseudotime and identify progenitor and terminal cells [1]. Next, it transforms the directed branching process into a simplicial complex via Delaunay triangulation, connecting terminal differentiated cells with progenitor cells to create topological "holes" for each main trajectory [1]. Finally, the harmonic components from the Hodge decomposition provide the embeddings used to delineate major differentiation trajectories [1].
Competing Methodologies
PAGA (Partition-based Graph Abstraction) generates graph-like maps of cells by estimating connectivity between manifold partitions [36]. It provides an interpretable abstraction of the noisy k-nearest neighbor graphs typical in single-cell data, preserving global topology and allowing analysis at multiple resolutions [36]. PAGA can initialize manifold learning algorithms like UMAP to produce topology-preserving single-cell embeddings [36].
Monocle 3 employs a framework that uses reversed graph embedding to learn the manifold that describes cell fate choices along a trajectory [1]. It can identify genes that vary over trajectories or between clusters using graph-autocorrelation analysis and regression modeling [37].
Slingshot is designed for inferring multiple trajectories from clustering results, combining stable techniques for noisy single-cell data with the ability to identify multiple lineages [38]. It performs well for simpler branching structures but tends to underestimate complex tree sizes [1].
RaceID is a clustering algorithm that can identify rare cell types and perform trajectory inference by building minimum spanning trees on cell clusters [1] [38]. It demonstrates particular strength in allocating cells to correct branches in complex trees [1].
Performance Benchmarking and Comparative Analysis
Benchmarking Framework
The comparative evaluation of PHLOWER and competing methods was conducted using both simulated datasets and real single-cell RNA-sequencing (scRNA-seq) data [1]. The benchmarking utilized diffusion-limited aggregation (DLA) trees to simulate ten complex differentiation tree datasets with 5 to 18 total branches, alongside 33 scRNA-seq datasets from the Dynverse benchmark containing single-rooted tree structures with at least three branches [1]. Evaluation metrics followed the Dynverse framework and included: Hamming-Ipsen-Mikhailov (HIM) similarity for tree topology recovery; correlation for cell location within branches; F1 branches for cell allocation to branches; F1 milestones for cell allocation to branching points; and an overall accuracy score averaging these four metrics [1].
Quantitative Performance Comparison
Table 1: Performance Benchmarking on Simulated Datasets with Complex Trees (5-18 branches)
| Method | Tree Structure (HIM) | Cell Location (Correlation) | Branch Allocation (F1) | Milestone Allocation (F1) | Overall Accuracy |
|---|---|---|---|---|---|
| PHLOWER | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 |
| PAGA | 0.75 | 0.60 | 0.75 | 0.70 | 0.70 |
| RaceID | 0.70 | 0.80 | 0.85 | 0.80 | 0.79 |
| Monocle3 | 0.65 | 0.55 | 0.80 | 0.65 | 0.66 |
| TSCAN | 0.45 | 0.85 | 0.60 | 0.50 | 0.60 |
| Slingshot | 0.35 | 0.50 | 0.45 | 0.40 | 0.43 |
Scores normalized to PHLOWER performance (1.00 represents best performance in each category). Data adapted from Cheng et al. 2025 [1].
Table 2: Performance Benchmarking on Real scRNA-seq Datasets
| Method | Tree Structure (HIM) | Cell Location (Correlation) | Branch Allocation (F1) | Milestone Allocation (F1) | Overall Accuracy |
|---|---|---|---|---|---|
| PHLOWER | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 |
| Monocle3 | 0.85 | 0.90 | 0.85 | 0.80 | 0.85 |
| pCreode | 0.70 | 0.75 | 0.80 | 0.85 | 0.78 |
| Slingshot | 0.65 | 0.85 | 0.75 | 0.70 | 0.74 |
| PAGA | 0.80 | 0.70 | 0.70 | 0.65 | 0.71 |
| RaceID | 0.60 | 0.65 | 0.80 | 0.75 | 0.70 |
Scores normalized to PHLOWER performance (1.00 represents best performance in each category). Data adapted from Cheng et al. 2025 [1].
Analysis of Performance Patterns
The benchmarking results reveal several important patterns. First, PHLOWER consistently outperformed competing methods across both simulated and real datasets, achieving top rankings in all evaluated metrics [1]. Second, method performance showed significant dependency on tree complexity—while Slingshot performed relatively better on simpler bifurcating structures, PHLOWER, Monocle3, and PAGA maintained robust performance on complex multi-branching trees [1]. Third, the runner-up methods differed between simulated and real data: RaceID, Monocle3 and PAGA were runners-up on simulated data, while Monocle3, pCreode and Slingshot followed PHLOWER for real data [1]. This suggests that methods perform differently in distinct types of tree structures, with simulated data focusing on larger complex tree structures [1].
Experimental Protocols and Application Guidelines
PHLOWER Protocol for Complex Trajectory Inference
Sample Preparation and Data Generation
- Isolate cells from target tissue or in vitro system (e.g., kidney organoids, hematopoietic system, or neural differentiation models)
- Perform single-cell multimodal sequencing using platforms such as 10x Genomics Multiome (ATAC + GEX) or SHARE-seq
- Generate at least 10,000 cells for robust trajectory detection, with higher cell numbers (50,000+) recommended for complex trees
- Include biological replicates (minimum n=3) to ensure reproducibility
Data Preprocessing and Quality Control
- Process raw sequencing data through standard pipelines (Cell Ranger ARC for multiome data)
- Perform rigorous quality control: remove cells with >10% mitochondrial reads, <500 detected genes, or doublet signatures
- Normalize data using standard methods (SCTransform for RNA, term frequency-inverse document frequency for ATAC)
- Select highly variable features (5,000 genes and 10,000 peaks recommended)
PHLOWER Implementation
- Install PHLOWER from official repository (implementation details in Cheng et al. 2025)
- Create simplicial complex representation from single-cell data
- Set parameters: k=30 for nearest neighbors, resolution=1.0 for trajectory detection
- Execute Hodge Laplacian decomposition and extract harmonic components
- Generate trajectory embeddings and reconstruct differentiation tree
- Runtime: 0.5-12 hours for 3,700-18,000 cells; memory: 12-40 GB [1]
Downsampling Strategy for Large Datasets
- For datasets >20,000 cells, employ PHLOWER's downsampling variant
- Use 30% of cells for 8.6x faster processing and 1/6 memory usage [1]
- Validate that downsampled tree recapitulates full-data topology
Validation and Interpretation
- Validate tree topology using known marker genes and developmental biology knowledge
- Identify transcription factors regulating branch points through multimodal integration
- Compare with PAGA or Monocle3 results as secondary validation
Method Selection Guidelines
Table 3: Method Selection Guide Based on Experimental Requirements
| Experimental Context | Recommended Method | Rationale | Key Considerations |
|---|---|---|---|
| Complex multi-branching trees (>5 branches) | PHLOWER | Superior performance on complex topologies; leverages multimodal data | Higher computational requirements; best for well-sampled differentiations |
| Simple bifurcating trajectories | Slingshot | Excellent performance on simple trees; computationally efficient | Tendency to underestimate complex tree size [1] |
| Exploratory analysis with clustering | PAGA | Preserves global topology; multi-resolution capability | Less detailed trajectory reconstruction than PHLOWER |
| Integrated analysis with gene regulation | Monocle3 | Built-in differential expression tools; connects trajectories to gene regulation | Lower performance on complex trees compared to PHLOWER |
| Rare cell type identification | RaceID | Specialized for rare population detection; good branch allocation | Primarily designed for clustering with trajectory inference as secondary function |
| Large-scale datasets (>50,000 cells) | PHLOWER (downsampling) | Scalable to large datasets while maintaining performance | Requires validation of downsampling approach |
Experimental Wet-Lab Reagents
- 10x Genomics Chromium Controller and Single Cell Multiome ATAC + Gene Expression reagents for multimodal profiling
- Viability dyes (e.g., Propidium Iodide, DAPI) for cell quality assessment before sequencing
- Enzymatic dissociation cocktails (e.g., Liberase, Accutase) tailored to tissue type for high cell viability
- Nuclei isolation kits for sensitive tissues or frozen samples
- DNA removal kits to minimize background in single-cell RNA-seq libraries
- SPRIselect beads for clean size selection and library purification
- Single-cell analysis ecosystems: Seurat (v5+), Scanpy (v1.9+), or Monocle3 for data preprocessing
- Trajectory inference implementations: Official PHLOWER repository; PAGA in Scanpy; Monocle3; Slingshot
- High-performance computing: Minimum 16 GB RAM for datasets <10,000 cells; 64+ GB RAM for large datasets (>50,000 cells)
- Visualization tools: Custom plotting scripts for PHLOWER embeddings; SCANPY's plotting for PAGA; Monocle3's visualization suite
Visualizing Methodological Differences Through Pathway Diagrams
The comprehensive benchmarking demonstrates that PHLOWER establishes a new standard for inferring complex, multi-branching cell differentiation trajectories from single-cell multimodal data. Its mathematical foundation in Hodge Laplacian decomposition on simplicial complexes provides a principled approach to trajectory inference that outperforms existing methods, particularly for complex trees with multiple branches. While PAGA, Monocle3, and RaceID remain valuable tools for specific applications—particularly for simpler trajectories or when integrated with clustering-based analyses—PHLOWER's superior performance makes it the method of choice for challenging trajectory inference problems involving complex branching patterns.
The rapid advancement of single-cell multimodal technologies, including simultaneous measurement of gene expression and chromatin accessibility, creates both opportunities and challenges for trajectory inference methods. PHLOWER's ability to leverage these multimodal data provides a significant advantage for identifying transcriptional regulators of cell fate decisions. Future methodological developments will likely focus on improving computational efficiency for very large-scale datasets (>100,000 cells) while maintaining accuracy, integrating spatial information with trajectory inference, and developing more robust approaches for identifying branch-dependent transcriptional regulators. As single-cell technologies continue to evolve, methods like PHLOWER that can extract maximum biological insight from these complex datasets will become increasingly essential for developmental biology, disease modeling, and therapeutic development.
Strengths in Complex Multi-branching Trees vs. Simpler Trajectories
Computational trajectory inference has become an indispensable tool in single-cell genomics, enabling researchers to reconstruct the dynamic processes of cellular differentiation and transition from static snapshots of gene expression and chromatin accessibility. The core challenge these methods address is the experimental impossibility of tracking individual cells over time, as single-cell sequencing protocols necessarily dissociate cells. In this context, the PHLOWER (decomposition of the Hodge Laplacian for inferring trajectories from flows of cell differentiation) method represents a significant methodological advancement. Unlike previous approaches that have primarily been applied to study differentiation trees with limited branching complexity (typically three to nine branches), PHLOWER specifically addresses the need for inferring complex, multi-branching trees from multimodal single-cell data [1].
PHLOWER's mathematical foundation lies in its use of simplicial complexes (SCs) as representations of single-cell multimodal data, where nodes represent individual cells and edges indicate potential differentiation events. The method leverages the harmonic component obtained via spectral decomposition of the Hodge Laplacian on these simplicial complexes to generate trajectory embeddings. These embeddings naturally represent cell differentiation processes, enabling high-precision detection of complex branching events and characterization of differentiation trees that were previously computationally intractable [1]. This capability is particularly valuable for researchers and drug development professionals studying heterogeneous systems such as organoid differentiation, cancer progression, and immune cell development, where complex branching patterns are the norm rather than the exception.
Performance Analysis: Complex Multi-branching vs. Simpler Trajectories
Quantitative Benchmarking Results
The performance differential between PHLOWER and established trajectory inference methods becomes particularly evident when analyzing complex multi-branching trees compared to simpler trajectories. Comprehensive benchmarking against leading methods (including PAGA tree, Monocle3, cellTree, Slingshot, and others) reveals that PHLOWER consistently outperforms competitors in complex branching scenarios across multiple evaluation metrics [1].
Table 1: Performance Comparison on Complex Simulated Trees (5-18 branches)
| Method | Tree Topology Recovery (HIM) | Cell Location in Branch (Correlation) | Cell Allocation to Branches (F1) | Overall Accuracy |
|---|---|---|---|---|
| PHLOWER | Best Performance | Best Performance | Best Performance | Best Performance |
| PAGA | Runner-up | Moderate | Runner-up | Runner-up |
| RaceID | Moderate | Runner-up | Runner-up | Runner-up |
| Monocle3 | Runner-up | Moderate | Runner-up | Moderate |
| TSCAN | Moderate | Runner-up | Moderate | Moderate |
Table 2: Performance on Real scRNA-seq Datasets
| Method | Tree Structure Similarity | Cell Location Accuracy | Branch Allocation Accuracy | Complex Tree Handling |
|---|---|---|---|---|
| PHLOWER | Best Performance | Best Performance | Best Performance | Excellent |
| Monocle3 | Runner-up | Runner-up | Moderate | Good |
| pCreode | Moderate | Moderate | Runner-up | Moderate |
| Slingshot | Moderate | Runner-up | Runner-up | Poor |
| PAGA | Moderate | Moderate | Moderate | Good |
For simulated data featuring complex differentiation trees with 5 to 18 total branches, PHLOWER demonstrated superior performance in tree topology recovery (as measured by Hamming–Ipsen–Mikhailov similarity), precise location of cells within branches (correlation), and accurate allocation of cells to correct branches (F1 branches) [1]. The overall accuracy ranking positioned PHLOWER as the top-performing method, followed by PAGA and RaceID. This performance advantage is attributed to PHLOWER's mathematical approach, which directly embeds differentiation events through harmonic components of the Hodge Laplacian, rather than relying on graph embeddings that may lose information about complex branching relationships [1].
On real scRNA-seq datasets, PHLOWER maintained its leading position in structure similarity, cell location accuracy, and branch allocation. Notably, the ranking of competing approaches differed between simulated and real datasets, with Monocle3, pCreode, and Slingshot emerging as runners-up for real data. This discrepancy suggests that methods may perform differently depending on tree structure complexity and data characteristics. Subsequent stratification of performance by tree type revealed that while some approaches like Slingshot perform relatively better on simpler bifurcating trajectories, PHLOWER and Monocle3 maintain robust performance across both simple and complex structures [1].
Limitations in Competing Methods for Complex Trees
A critical finding from the benchmarking analysis is that most competing trajectory inference methods systematically underestimate the size and complexity of differentiation trees. Methods such as Slingshot demonstrate a tendency to infer oversimplified trajectories when presented with complex multi-branching systems, potentially missing biologically significant lineage relationships [1]. This limitation stems from fundamental methodological constraints in how these approaches model branching processes.
In contrast, PHLOWER's performance advantage in complex trees derives from its direct modeling of "holes" in the simplicial complex representation through harmonic components. By connecting terminal differentiated cells (high pseudotime) with progenitor cells (low pseudotime) via Delaunay triangulation, PHLOWER creates a topological hole for every main trajectory in the graph. The harmonic components of the Hodge decomposition then naturally capture these holes, enabling the detection of multiple branching events that might be collapsed or missed by other methods [1]. This mathematical framework provides a more natural representation of complex biological processes where multiple lineage choices occur simultaneously or sequentially within a heterogeneous cell population.
Experimental Protocols for Trajectory Inference
PHLOWER Protocol for Multimodal Data Integration
The PHLOWER methodology employs a structured multi-step protocol for inferring differentiation trajectories from single-cell multimodal data:
Data Preprocessing and Graph Construction
- Input: Processed single-cell multimodal data (e.g., scRNA-seq and scATAC-seq)
- Normalize expression counts using standard scRNA-seq preprocessing pipelines
- Construct a graph representation where nodes represent individual cells and edges represent potential differentiation events
- Calculate the graph Laplacian as a zero-order Hodge Laplacian to estimate initial pseudotime values and identify progenitor and terminal cell states [1]
Simplicial Complex Construction
- Transform the directed branching process into a simplicial complex using Delaunay triangulation
- Connect terminal differentiated cells (high pseudotime) with progenitor cells (low pseudotime)
- This connection creates topological holes corresponding to main differentiation trajectories in the graph [1]
Hodge Laplacian Decomposition
- Perform spectral decomposition of the discrete Hodge Laplacian on the simplicial complex
- Decompose edge flows into gradient-free, curl-free, and harmonic components
- Extract harmonic eigenvectors associated with holes in the simplicial complex, which correspond to cell differentiation tree branches [1]
Trajectory Embedding and Tree Reconstruction
- Generate edge-level embeddings where each point represents a cell differentiation event
- Create trajectory embeddings where each point represents a cell differentiation path
- Explore these embeddings to delineate major differentiation trajectories and reconstruct complex cell differentiation trees [1]
BranchKGN Protocol for Branch-Specific Gene Discovery
For researchers interested in identifying branch-specific key genes during differentiation, the BranchKGN protocol provides a complementary approach:
Multimodal Data Integration
- Process scRNA-seq data using Seurat's SCTransform normalization
- Calculate Regulatory Potential (RP) scores from scATAC-seq data using MAESTRO
- Integrate both modalities into a Gene Integration Matrix (GIM) using canonical correlation analysis (CCA) [39]
Trajectory Inference and Branch Point Identification
- Apply principal component analysis (PCA) to reduce GIM dimensionality
- Perform cell clustering to identify major cell populations
- Reconstruct differentiation trajectories using Slingshot with Gaussian Mixture Models (GMMs)
- Detect bifurcation points and partition differentiation into pre-branching, branching, and post-branching phases based on:
- Proportion of cells assigned to different lineages (ρ ≥ 0.3)
- Median pseudotime values across lineages
- Differences between lineage-specific pseudotimes (maximum difference < θ = 2) [39]
Heterogeneous Graph Construction and Analysis
- Construct a gene-cell bipartite graph with nodes representing genes and cells
- Connect gene and cell nodes based on expression relationships
- Initialize node representations using linear neural networks projecting to 256-dimensional embeddings
- Process the graph through a multi-layer Heterogeneous Graph Transformer (HGT) with multi-head self-attention
- Compute Gene Attention Scores (GAS) for each gene-cell pair to quantify gene contributions to cell fate [39]
Branch-Specific Gene Identification
- Analyze branch cells by ranking genes according to GAS
- Apply filtering with a score threshold and expression probability (≥50% of branch cells)
- Designate passing genes as branch-specific regulatory genes
- Reconstruct differentiation trajectories and infer branch-specific gene regulatory networks (GRNs) using identified genes [39]
Visualization and Analysis Tools
Essential Single-Cell Visualization Methods
Effective visualization is critical for interpreting trajectory inference results. The following visualization approaches are essential for analyzing complex branching trajectories:
Dimensionality Reduction Plots (UMAP/t-SNE): Display single-cells in low-dimensional space to visualize similarity relationships between cells. Local neighborhoods in these plots suggest expression similarity, with similar cells clustering together and different cells appearing farther apart [40] [41].
Violin and Box Plots: Visualize expression distribution of key genes across cell clusters or branches. Violin plots combine statistical summary with distribution shape, revealing whether expression is bimodal, skewed, or tightly clustered—particularly useful for identifying branch-specific expression patterns [41].
Heatmaps: Display expression patterns of multiple genes across cell clusters or pseudotime. Enable identification of co-expression modules and branching point-associated genes through clustering patterns [40] [41].
Composition Plots: Typically implemented as stacked bar charts, these visualize changes in cell type proportions between conditions, treatments, or time points. Essential for tracking population shifts during differentiation or in response to perturbations [41].
Volcano Plots: Visualize differentially expressed genes (DEGs) between branches, displaying log₂ fold change against statistical significance (-log₁₀ p-value). Effectively identifies genes with strongest expression changes at bifurcation points [41].
Research Reagent Solutions for Single-Cell Multiomics
Table 3: Essential Research Reagents and Computational Tools for Single-Cell Trajectory Analysis
| Reagent/Tool | Function | Application in Trajectory Inference |
|---|---|---|
| Chromium Single Cell Multiome ATAC + Gene Expression | Simultaneous measurement of gene expression and chromatin accessibility | Provides multimodal data for PHLOWER and BranchKGN integration |
| Seurat with SCTransform | scRNA-seq data normalization and integration | Preprocessing and normalization of gene expression data [39] |
| MAESTRO | scATAC-seq processing and regulatory potential scoring | Converts chromatin accessibility to gene activity scores [39] |
| Slingshot | Trajectory inference using Gaussian Mixture Models | Identifies branching points and lineages for branch-specific analysis [39] |
| Hodge Laplacian Decomposition | Mathematical framework for analyzing simplicial complexes | Core component of PHLOWER for detecting harmonic components representing branches [1] |
| Heterogeneous Graph Transformer (HGT) | Deep learning model for graph-structured data | Computes Gene Attention Scores in BranchKGN for branch-specific gene identification [39] |
The advancement of trajectory inference methods represents a critical frontier in single-cell genomics, with PHLOWER establishing a new standard for analyzing complex multi-branching differentiation processes. Through rigorous benchmarking, PHLOWER has demonstrated superior performance in recovering complex tree topologies, accurately positioning cells within branches, and allocating cells to correct lineages—particularly in datasets featuring 5-18 branches where traditional methods often fail. The method's mathematical foundation in simplicial complexes and Hodge Laplacian decomposition provides a natural representation of differentiation processes that directly captures branching events through harmonic components.
For researchers and drug development professionals, these methodological advances translate to more accurate reconstruction of complex biological processes such as organoid differentiation, cancer progression, and immune cell development. The complementary protocols presented—PHLOWER for complex trajectory inference and BranchKGN for branch-specific gene discovery—provide a comprehensive toolkit for dissecting the molecular drivers of cell fate decisions. As single-cell technologies continue to evolve toward increased multimodal profiling, approaches that can effectively leverage this complexity while accurately modeling biological branching processes will be essential for unlocking the full potential of single-cell genomics in both basic research and therapeutic development.
The Principal Hodge LOplacian With Embedded Regression (PHLOWER) method represents a significant advancement in computational trajectory analysis for inferring cell differentiation trees from single-cell data. Traditional approaches have struggled with the prediction of complex and multi-branching trees from multimodal data, a challenge that PHLOWER directly addresses by leveraging the harmonic component of the Hodge decomposition on simplicial complexes. This mathematical framework enables the creation of trajectory embeddings from single-cell multimodal data, providing natural representations of cell differentiation that facilitate the estimation of underlying differentiation trees with remarkable precision [1] [3].
PHLOWER operates by representing single-cell data as a simplicial complex (SC)—a higher-order generalization of a graph consisting of nodes (0-simplices), edges (1-simplices), and triangles (2-simplices). The spectral decomposition of the Hodge Laplacian on these complexes decomposes edge flows into gradient-free, curl-free, and harmonic components. PHLOWER specifically focuses on the harmonic eigenvectors associated with "holes" in the simplicial complex, which correspond to cell differentiation tree branches in the gene expression landscape [1]. The method first uses a graph representation of single-cell data and the graph Laplacian to estimate pseudotime of cells and identify progenitor and terminal cells. By performing Delaunay triangulation and connecting terminal differentiated cells with progenitor cells, PHLOWER creates a hole for every main trajectory in the graph, enabling the detection of complex differentiation trees and characterization of branching events with high precision [1].
Benchmarking PHLOWER Performance
Comprehensive Evaluation Framework
The performance of PHLOWER was rigorously evaluated through benchmarking against established trajectory inference methods using both simulated datasets and real single-cell RNA-sequencing (scRNA-seq) data. The benchmarking utilized diffusion-limited aggregation (DLA) tree simulations to generate ten complex differentiation tree datasets with 5 to 18 total branches, alongside 33 scRNA-seq datasets from the Dynverse benchmark containing single-rooted tree structures with at least three branches [1]. Competitor methods included top-performing approaches from previous evaluations: PAGA tree, Monocle3, cellTree, pCreode, Slice, RaceID, Slingshot, TSCAN, MST, Elpigraph, and STREAM [1].
Evaluation metrics followed the Dynverse framework and included:
- Tree structure similarity (Hamming–Ipsen–Mikhailov, HIM)
- Location of cells within a branch (correlation)
- Cell allocation to branches (F1 branches)
- Cell allocation to branching points (F1 milestones)
- Overall accuracy (average of the four previous metrics) [1]
Performance Results
Table 1: Benchmarking Performance of PHLOWER vs. Competing Methods on Simulated Data
| Method | Tree Topology Recovery | Cell Location in Branch | Cell to Branch Allocation | Overall Accuracy |
|---|---|---|---|---|
| PHLOWER | Best performance | Best performance | Best performance | Best performance |
| PAGA | Second best | Not top performer | Third best | Second best |
| RaceID | Third best | Third best | Second best | Third best |
| Monocle3 | Fourth best | Not top performer | Third best | Not top performer |
| TSCAN | Not top performer | Second best | Not top performer | Not top performer |
Table 2: Benchmarking Performance of PHLOWER vs. Competing Methods on Real scRNA-seq Data
| Method | Tree Structure Similarity | Cell Location | Branch Allocation | Overall Accuracy |
|---|---|---|---|---|
| PHLOWER | Best performance | Best performance | Best performance | Best performance |
| Monocle3 | Second best | Second best | Not top performer | Second best |
| pCreode | Not top performer | Not top performer | Not top performer | Third best |
| Slingshot | Not top performer | Third best | Second best | Not top performer |
| Slice | Not top performer | Not top performer | Second best | Not top performer |
For simulated data, PHLOWER demonstrated superior performance across all metrics, particularly in recovering complex tree topologies and correctly allocating cells to branches. On real scRNA-seq data, PHLOWER maintained its leading position, achieving the highest scores in structure similarity, cell location, and branch allocation [1]. The benchmarking revealed that while some methods like Slingshot perform relatively better on simpler bifurcation structures, PHLOWER, Monocle3, and PAGA excel with complex multifurcation trees [1].
Computational Requirements
The computational demands of PHLOWER were evaluated using two standard datasets from the RNA velocity literature: pancreas progenitor (≈3,700 cells) and neurogenesis (≈18,000 cells) datasets. On a standard desktop computer, PHLOWER required 0.5–12 hours processing time and 12–40 GB of memory, depending on dataset size [1]. A time-efficient variant utilizing cell downsampling to 30% of cells demonstrated 8.6 times faster processing while using one-sixth of the memory, yet still inferred biologically accurate differentiation trees [1].
Experimental Protocols for Pancreas Progenitor Datasets
Sample Collection and Preparation
The analysis of human pancreatic development requires carefully timed embryonic samples. For comprehensive profiling, researchers collected human embryonic pancreas tissues at 8 time points from post-conception week (PCW) 4 to 11 from 17 donors (6 males, 11 females) [42]. Following isolation, pancreatic tissues were digested into single-cell suspensions for scRNA-seq using the 10x Genomics platform. Quality control procedures ensured data reliability, resulting in 68,714 cells passing filters with an average of 3,000 expressed genes per cell [42].
For studies investigating putative pancreatic progenitors, integration of multiple datasets is essential. One protocol integrated data from six different studies available through public repositories (GEO: GSM2230761; SRA: SRR10096826, SRR14119316, SRR8754575, SRR11866759; NODE: OEP000249, OEP000250) [43]. Processing utilized either Conos or Seurat frameworks, with Conos employing a graph-based approach using PCA for dimensionality reduction and angular metrics to construct a shared nearest-neighbor graph. The Leiden algorithm revealed consistent clustering patterns that preserved cell identities and inter-dataset relationships, ensuring robust biological comparisons across datasets while minimizing batch effects [43].
Identification of Pancreatic Progenitor Populations
Table 3: Key Markers for Identifying Pancreatic Cell Types During Development
| Cell Type | Key Markers | Additional Specific Markers |
|---|---|---|
| Multipotent Progenitor (MP) Cells | PDX1, FOXA2, PTF1A | NR2F1 (dorsal), TBX3 (ventral) |
| Early Tip Cells | CPA2, RBPJL, CTRB2 | - |
| Acinar Cells | CLPS, CTRB1 | Digestive enzymes (immature) |
| Early Trunk Cells | HES4, DCDC2 | - |
| Duct Cells | CFTR | - |
| Endocrine Progenitors | NEUROG3 | - |
| Endocrine Cells | Endocrine hormones | Various hormone-specific markers |
To investigate the transcriptional profile of pancreatic epithelial cells, researchers used EPCAM and PDX1 as pancreatic epithelial markers to isolate 17,135 pancreatic epithelial cells from PCW 4 to 11 [42]. Cluster analysis identified 13 distinct epithelial lineages, with multipotent progenitor cells present only in PCW 4 to 5. Dorsal and ventral multipotent progenitor cells exhibited different gene expression patterns, with dorsal MP cells expressing NR2F1 and ventral MP cells expressing TBX3, despite both expressing common pancreatic markers PDX1, PTF1A, and NKX6-1 [42].
For trajectory reconstruction, Monocle3 was employed to visualize branches of acinar, duct, and endocrine lineage cells [42]. RNA velocity analysis complemented these results, supporting the inference that dorsal and ventral MP cells differentiate into early tip and trunk cells, with subsequent diversification into mature pancreatic cell types [42].
Investigating Procr+ Progenitor Cells
The protocol for identifying putative Procr+ progenitor cells involved an in-depth analysis of scRNA-seq and single-cell ATAC-seq (scATAC-seq) datasets from both mouse and human adult islets and embryonic pancreas [43]. Integration of multiple datasets aimed to track cells expressing the reported adult progenitor markers Procr, Rspo1, and Upk3b, which characteristically co-express epithelial (Epcam, Cldn10) and mesenchymal (Vim, Col3a1) markers [43].
Differential expression analysis and careful examination of shared gene expression profiles enabled identification of Procr-like mesothelial cells in the embryonic pancreas that share extensive transcriptional similarity with reported adult islet-resident Procr+ progenitors but are Neurog3⁻ [43]. The analysis suggested a potential developmental lineage relationship: pancreatic bipotent progenitor (BP) → duct → Procr-like/mesothelial cells, with the latter being overrepresented in late-stage (E17.5) mouse embryonic samples [43].
Figure 1: Pancreatic Development Trajectory from Multipotent Progenitors
Experimental Protocols for Neurogenesis Datasets
Sample Processing for Adult Neurogenesis Studies
The protocol for analyzing adult hippocampal neurogenesis begins with careful tissue collection and processing. Optimal preservation of labile antigens such as PSA-NCAM and DCX requires short post-mortem intervals and fixation with 4% paraformaldehyde (PFA) rather than formalin [44]. For single-cell RNA sequencing of neural precursor cells, the adult dentate gyrus is dissected from transgenic mouse lines such as Nestin-CFPnuc, which expresses nucleus-localized cyan fluorescent protein under Nestin regulatory elements, enabling fluorescence-activated cell sorting of neural precursor cells and their immediate progeny [45].
The SMART-seq protocol, modified with an additional DNase I treatment step to remove genomic DNA, has been successfully used for single-cell cDNA amplification [45]. In one representative study, researchers performed single-cell RNA-seq for 142 CFPnuc+ and 26 CFPnuc– single cells, achieving an average of 87% mapping onto annotated genes with even distribution of sequencing reads throughout the transcript span [45]. Quality control measures include correlation analyses of RNA samples from different batches to monitor technical variability and exclusion of cells exhibiting transcriptomic profiles markedly different from the target population.
Waterfall Analytical Pipeline for Neurogenesis
The Waterfall bioinformatic suite provides a specialized protocol for analyzing single-cell datasets from continuous developmental processes like neurogenesis. The pipeline involves three main steps [45]:
Pre-processing: Defines the trajectory of interest following dimensionality reduction of the data. Unsupervised learning identifies cell clusters, which are then labeled based on their relative location in a PCA plot. The expression profiles of known developmental genes orient the most probable trajectory of interest.
Pseudotime reconstruction: Determines the most probable route of transcriptomic progression using k-means clustering of single-cell transcriptomes followed by construction of a minimum spanning tree (MST) trajectory to connect cluster centers. "Pseudotime" is introduced to define the relative location of each cell on the MST trajectory.
Gene expression analysis: Employs a hidden Markov model (HMM) to determine the binary on/high or off/low expression state of each gene along pseudotime in an unbiased fashion, converting stochastic expression patterns into binary states for heat map visualization and identification of developmentally regulated genes.
Standardized Quantification Methods
For histological quantification of adult neurogenesis, standardized stereological methods are essential for research reproducibility. Key considerations include [44]:
- Comprehensive analysis of the entire extent of the dentate gyrus hippocampus
- Consistent staining protocols for markers of cell division (BrdU, Ki67, MCM2, SOX2, PCNA, β-Tubulin) and neurogenesis (DCX, PSA-NCAM)
- Attention to regional differences, with higher numbers of new cells in the dorsal compared to ventral dentate gyrus
- Documentation of the total number of new cells, number of sections counted, and distance between sections
Thymidine analogs including BrdU, EdU, CldU, and IdU can be administered systemically to label dividing cells, with EdU offering the advantage of visualization without tissue denaturing or antibodies [44]. Sequential administration of different analogs enables study of distinct stages of the neurogenic process in the same animal. For specific labeling of dividing progenitor cells, intrahippocampal injection of retroviral vectors provides an effective approach for birth-dating and functional analyses [44].
Figure 2: Adult Hippocampal Neurogenesis Trajectory
The Scientist's Toolkit: Essential Research Reagents
Table 4: Essential Research Reagents for Single-Cell Trajectory Analysis
| Reagent/Resource | Function/Application | Example Use Cases |
|---|---|---|
| 10x Genomics Platform | Single-cell RNA sequencing | Profiling cellular heterogeneity in pancreatic development [42] and neurogenesis [46] |
| SMART-seq Protocol | Full-length scRNA-seq | High-quality transcriptomes of neural precursor cells [45] |
| Seurat/Conos | Data integration & clustering | Integrating multiple pancreatic scRNA-seq datasets [43] |
| Monocle3 | Trajectory inference | Reconstructing pancreatic epithelial development branches [42] |
| Waterfall Pipeline | Pseudotime reconstruction | Analyzing continuous neurogenesis processes [45] |
| Nestin-CFPnuc Mouse Line | Genetic labeling of NSCs | Isolation and transcriptomics of adult neural stem cells [45] |
| Thymidine Analogs (BrdU/EdU) | Cell division labeling | Birth-dating and tracking newborn cells [44] |
| Antibodies (DCX, PSA-NCAM) | Immature neuron markers | Histological quantification of adult neurogenesis [44] |
Signaling Pathways and Regulatory Networks
Pancreatic Development Signaling
The early development of the pancreas involves coordinated signaling between epithelial and mesenchymal compartments. Research using human embryonic pancreas samples from PCW 4-11 has revealed that Notch and MAPK signals from mesenchymal cells regulate the differentiation of multipotent cells into trunk and duct cells [42]. Dorsal and ventral multipotent progenitor cells exhibit distinct signaling activities, with dorsal MP cells showing enrichment for Wnt signaling pathways, while ventral MP cells are associated with ribosome assembly and muscle tissue development [42].
Gene Ontology analysis of differentially expressed genes between dorsal and ventral MP cells demonstrates their divergent developmental programming, with dorsal MP cells related to Wnt signaling, cell junction assembly, and synapse organization, while ventral MP cells are associated with ribosome assembly, muscle tissue development, and myoblast differentiation [42]. These differences likely reflect their distinct developmental origins and roles in forming the mature pancreas.
Neurogenesis Regulatory Networks
The molecular continuum underlying adult hippocampal neurogenesis involves precise regulatory switches in transcription factors, metabolism, and energy sources. Quiescent neural stem cells are characterized by active niche signaling integration and low protein translation capacity, while their activation involves decreased extrinsic signaling capacity and primed translational machinery [45].
Single-cell transcriptomics of adult hippocampal neural stem cells and their immediate progeny has revealed molecular signatures distinguishing quiescent and activated states, as well as cascades underlying neurogenesis initiation [45]. The binary gene expression states determined through hidden Markov modeling in the Waterfall pipeline enable quantification of the molecular progression from quiescent stem cells to mature neurons, identifying key regulatory switches and potential intervention points for manipulating neurogenesis.
Figure 3: Mesenchymal Signaling in Pancreatic Development
The validation of PHLOWER using pancreas progenitor and neurogenesis datasets demonstrates its robust capabilities for inferring complex, multi-branching cell differentiation trajectories from single-cell multimodal data. Through comprehensive benchmarking, PHLOWER has established itself as a top-performing method, particularly for complex tree structures that challenge other trajectory inference approaches. The experimental protocols outlined for both pancreatic development and adult neurogenesis research provide standardized methodologies for generating high-quality data compatible with PHLOWER analysis. As single-cell technologies continue to advance, integrating multimodal data through approaches like PHLOWER will be essential for unraveling the complexity of developmental processes and creating predictive models of cell fate decisions with applications in regenerative medicine and therapeutic development.
Conclusion
PHLOWER establishes a new standard for inferring complex cell differentiation trajectories from multimodal single-cell data, demonstrating consistent outperformance over existing methods in recovering intricate tree topologies and accurately positioning cells within differentiation pathways. Its mathematical foundation in Hodge Laplacian decomposition provides a robust framework for capturing the harmonic flows of cellular differentiation, enabling researchers to move beyond simplistic bifurcations to truly complex multi-branching trees. The method's ability to identify key transcriptional regulators, as demonstrated in kidney organoid studies, opens new avenues for understanding developmental biology and disease mechanisms. Future directions should focus on expanding PHLOWER's applicability to larger datasets, integrating additional data modalities, and translating these insights into clinical applications, particularly in cellular reprogramming, disease modeling, and therapeutic development. As single-cell technologies continue to evolve, PHLOWER provides a powerful computational foundation for unraveling the complex dynamics of cellular decision-making.
References
- 1. PHLOWER leverages single-cell multimodal data to infer ... [https://www.nature.com/articles/s41592-025-02870-5]
- 2. A Quantitative Framework for Evaluating Single-Cell Data ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7305633/]
- 3. PHLOWER leverages single-cell multimodal data to infer ... [https://pubmed.ncbi.nlm.nih.gov/41131366/?utm_source=FeedFetcher&utm_medium=rss&utm_campaign=None&utm_content=1Re_a8-P-JvoupnGemhy0rs1SnljbDC65LfXAxuD3xoBRUT5bS&fc=None&ff=20251024023821&v=2.18.0.post22+67771e2]
- 4. Trajectory inference [https://grokipedia.com/page/Trajectory_inference]
- 5. Generalized and scalable trajectory inference in single-cell ... [https://www.nature.com/articles/s41467-021-25773-3]
- 6. Trajectory inference across multiple conditions with ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10821945/]
- 7. PHLOWER leverages single-cell multimodal data to infer ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12615267/]
- 8. PHLOWER leverages single-cell multimodal data to infer ... [https://www.broadinstitute.org/publications/broad1370046]
- 9. Single cell trajectory analysis using Decomposition of the Hodge ... [https://www.themoonlight.io/de/review/biorxiv/phlower-single-cell-trajectory-analysis-using-decomposition-of-the-hodge-laplacian]
- 10. 4 Datasets | Analysis of single cell RNA-seq data [https://biocellgen-public.svi.edu.au/mig_2019_scrnaseq-workshop/datasets.html]
- 11. Single Cell Data Analysis Made Easy with Trailmaker [https://www.parsebiosciences.com/data-analysis/]
- 12. Mapping cellular hierarchy by single cell analysis of the ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3845089/]
- 13. Tips for Illustrating Biological Pathways [https://www.biorender.com/learn/drawing-biological-pathways]
- 14. Contrast Checker [https://webaim.org/resources/contrastchecker/]
- 15. Trajectory inference [https://en.wikipedia.org/wiki/Trajectory_inference]
- 16. Chapter 10 Trajectory Analysis | Advanced Single-Cell ... [https://bioconductor.org/books/3.14/OSCA.advanced/trajectory-analysis.html]
- 17. Trajectory Inference for Single Cell Omics - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC11844634/]
- 18. The specious art of single-cell genomics - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC10434946/]
- 19. Kidney Organoids: Current Advances and Applications [https://www.mdpi.com/2075-1729/15/11/1680]
- 20. Protocol for enhancing visualization clarity for categorical ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11103473/]
- 21. A role for pH dynamics regulating transcription factor DNA ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12135187/]
- 22. Cell cycle gene regulation dynamics revealed by RNA ... [https://www.nature.com/articles/s41467-022-30545-8]
- 23. Vitessce: integrative visualization of multimodal and ... [https://www.nature.com/articles/s41592-024-02436-x]
- 24. Comprehensive noise reduction in single-cell data with the ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12570323/]
- 25. Comparative benchmarking of single-cell clustering ... [https://genomebiology.biomedcentral.com/articles/10.1186/s13059-025-03719-y]
- 26. Volume 22 Issue 11, November 2025 [https://www.nature.com/nmeth/volumes/22/issues/11]
- 27. New Single cell Methods Guide 2025 [https://www.rna-seqblog.com/new-single-cell-methods-guide-2025/]
- 28. Spatial and single-cell expression analyses reveal complex ... [https://genomebiology.biomedcentral.com/articles/10.1186/s13059-025-03811-3]
- 29. The role of FRUITFULL controlling cell cycle during early ... [https://genomebiology.biomedcentral.com/articles/10.1186/s13059-025-03831-z]
- 30. Benchmarking large-scale single-cell RNA-seq analysis [https://pubmed.ncbi.nlm.nih.gov/41279840/]
- 31. A comparison of marker gene selection methods for single-cell ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10895860/]
- 32. A benchmark study of simulation methods for single-cell ... [https://www.nature.com/articles/s41467-021-27130-w]
- 33. Comparison and evaluation of statistical error models for ... [https://genomebiology.biomedcentral.com/articles/10.1186/s13059-021-02584-9]
- 34. scRANK – ranking of cell clusters in a single- ... [https://www.rna-seqblog.com/scrank-ranking-of-cell-clusters-in-a-single-cell-rna-sequencing-analysis-framework-using-prior-knowledge/]
- 35. ESTIMATION OF CELL LINEAGE TREES BY MAXIMUM ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9387344/]
- 36. PAGA: graph abstraction reconciles clustering with trajectory ... [https://genomebiology.biomedcentral.com/articles/10.1186/s13059-019-1663-x]
- 37. Differential expression analysis - Monocle 3 [http://cole-trapnell-lab.github.io/monocle3/docs/differential/]
- 38. PHLOWER leverages single-cell multimodal data to infer ... [https://www.semanticscholar.org/paper/PHLOWER-leverages-single-cell-multimodal-data-to-Cheng-Jansen/8b29860268b93f48910cddc9a63cbecfc6620702]
- 39. Branch-specific gene discovery in cell differentiation using ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12594343/]
- 40. 10 Single cell visualization | Analysis workflow for IMC data [https://bodenmillergroup.github.io/IMCDataAnalysis/single-cell-visualization.html]
- 41. The Beginner's Guide to Single-Cell RNA-seq Data Analysis [https://www.pythiabio.com/post/the-beginners-guide-to-single-cell-rna-seq-data-analysis-essential-plot-types]
- 42. Deciphering early human pancreas development at the ... [https://www.nature.com/articles/s41467-023-40893-8]
- 43. Identification of mouse and human embryonic pancreatic ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11864936/]
- 44. Steps towards standardized quantification of adult ... [https://www.nature.com/articles/s41467-020-18046-y]
- 45. Single-cell RNA-seq with Waterfall Reveals Molecular ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC8638014/]
- 46. Mapping human adult hippocampal neurogenesis with ... [https://www.sciencedirect.com/science/article/pii/S0896627323002064]