Deep Homology in Animal Design: From Evolutionary Concepts to Drug Discovery Applications
This article explores the concept of deep homology—the remarkable conservation of genetic regulatory circuits across distantly related animal species—and its profound implications for biomedical research.
Deep Homology in Animal Design: From Evolutionary Concepts to Drug Discovery Applications
Abstract
This article explores the concept of deep homology—the remarkable conservation of genetic regulatory circuits across distantly related animal species—and its profound implications for biomedical research. We first establish the foundational principles of deep homology, tracing its origins in evolutionary developmental biology (evo-devo) and its distinction from traditional homology. The discussion then progresses to methodological advances, including next-generation sequencing and protein language models like DHR, that enable the detection of deeply homologous systems. For the practicing researcher, we address common challenges in translating these concepts, such as animal model selection and statistical validation, providing optimization strategies. Finally, we present a comparative analysis of how deep homology informs target prioritization and structure-based drug design, validating its utility across pharmaceutical applications. This synthesis provides drug development professionals with a comprehensive framework for leveraging evolutionary conservation in therapeutic innovation.
The Evolutionary Blueprint: Uncovering Deep Homology in Animal Design
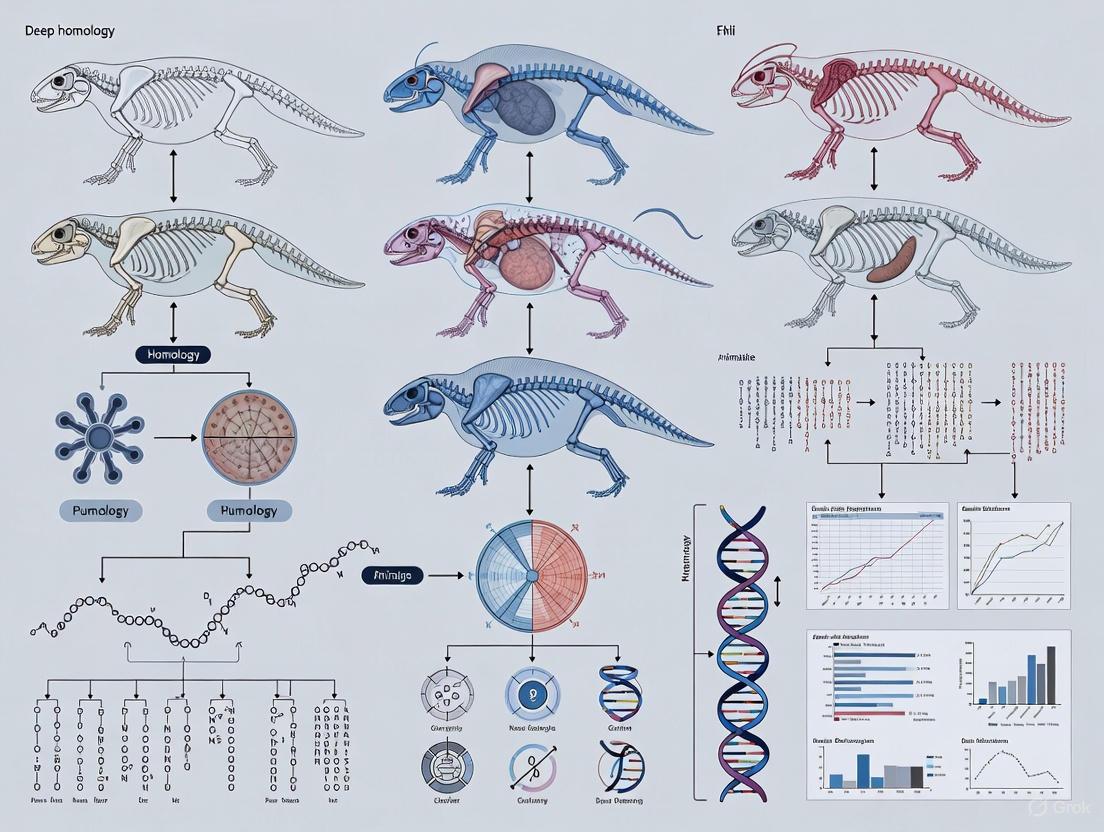
Deep homology represents a foundational concept in evolutionary developmental biology (evo-devo), describing the phenomenon where distantly related organisms share genetic regulatory apparatus used to build morphologically distinct and phylogenetically separate anatomical features. This conceptual framework has transformed our understanding of how evolutionary novelty is generated, revealing that conserved genetic toolkits are redeployed across deep evolutionary time. The intellectual journey of deep homology stretches from nineteenth-century anatomical theories to contemporary molecular genetics, creating a continuous thread in biological thought.
The significance of deep homology extends beyond academic evolutionary biology into practical biomedical applications. By revealing deeply conserved genetic pathways, it provides models for understanding human development and disease. For drug development professionals, these conserved pathways offer potential therapeutic targets and model systems for investigating disease mechanisms. This technical guide explores the conceptual, historical, and methodological evolution of deep homology, providing researchers with both theoretical framework and practical experimental approaches for contemporary investigations.
Historical Foundations: From Owen's Archetype to Darwinian Evolution
Richard Owen and the Vertebral Archetype
The intellectual precursor to deep homology emerged in the work of Victorian anatomist Sir Richard Owen (1804-1892), who introduced the concept of the archetype—a fundamental structural plan underlying anatomical diversity. In his 1848 work On the Archetype and Homologies of the Vertebrate Skeleton, Owen defined two critical anatomical relationships that would inform future homology concepts [1]:
- Homologue: "The same organ in different animals under every variety of form and function"
- Analogue: "A part or organ in one animal having the same function as another part or organ in a different animal"
Owen's vertebrate archetype represented an idealized primitive pattern—a generalized segmental design—from which all vertebrate skeletons could be derived. This Platonic conception viewed the archetype as an abstract blueprint existing in nature, with actual vertebrate skeletons representing variations on this theme [1]. His theory constituted a comprehensive synthesis of paleontology, comparative anatomy, and Christian Platonism, representing the culmination of typological thinking in biology.
Conceptual Transition to Evolutionary Homology
Charles Darwin's theory of evolution by natural selection transformed Owen's archetype from an abstract ideal to a historical ancestor—with the archetype reconceptualized as the common ancestor of the vertebrate lineage. This Darwinian reinterpretation maintained the concept of structural unity but provided a mechanistic, historical explanation rather than an idealist one.
The late 20th century saw the emergence of evolutionary developmental biology (evo-devo), which integrated comparative embryology, molecular genetics, and evolutionary theory. This synthesis set the stage for the modern conception of deep homology by focusing on the evolutionary modifications of developmental processes [2].
Table 1: Key Historical Concepts in the Development of Deep Homology
| Concept | Key Proponent | Time Period | Core Idea |
|---|---|---|---|
| Archetype | Richard Owen | 1840s | Ideal structural plan underlying anatomical diversity |
| Homology vs. Analogy | Owen | 1840s | Distinction between structural equivalence versus functional similarity |
| Descent with Modification | Charles Darwin | 1859 | Evolutionary transformation of ancestral structures |
| Genetic Toolkit | Evo-devo researchers | 1990s | Conserved genes regulating development across phylogeny |
| Deep Homology | Neil Shubin et al. | 2000s | Shared genetic regulatory apparatus underlying analogous features |
Modern Conceptual Framework: Principles and Mechanisms
Defining Deep Homology in Contemporary Terms
Deep homology extends beyond traditional morphological homology by revealing that distantly related lineages share genetic regulatory mechanisms that control the development of analogous structures. Unlike standard homology (which describes structures inherited from a common ancestor) or convergence (similar features arising independently), deep homology represents the independent co-option of homologous genetic circuits to build what become anatomically distinct features [3].
The core principle recognizes that while the morphological structures themselves may not be homologous (in the traditional sense of shared ancestry), the genetic regulatory networks that pattern their development are homologous and have been conserved over vast evolutionary time [3]. This represents a paradigm shift from comparing anatomical structures to comparing the genetic and developmental processes that generate those structures.
Mechanisms of Deep Homology
Several evolutionary mechanisms enable the conservation and redeployment of genetic toolkits across deep evolutionary distances:
- Conserved transcription factors: Regulatory proteins like Pax6, FoxP2, and Hox genes maintain their regulatory functions across animal phylogeny, despite extensive sequence divergence in target genes [3]
- Heterotopy: Spatial changes in gene expression patterns allow homologous genes to be deployed in novel developmental contexts
- Heterochrony: Temporal shifts in gene expression timing enable the modification of developmental trajectories
- Network co-option: Entire genetic modules are recruited for new developmental functions while maintaining their core regulatory logic
The molecular analysis of behavioral traits, including the role of FoxP2 in vocal learning across humans and songbirds, exemplifies how deep homology extends beyond morphology to complex behaviors [3].
Case Studies in Deep Homology: Empirical Evidence
Limb Development in Vertebrates and Insects
One of the most compelling examples of deep homology comes from the genetic regulation of appendage development across phyla. The Distal-less (Dll/Dlx) gene family, which patterns limb outgrowth in both vertebrates and insects, demonstrates how conserved genetic toolkits regulate the development of phylogenetically separate structures [3]. Despite the independent evolutionary origins of vertebrate and arthropod limbs, they share fundamental genetic patterning mechanisms.
Eye Development Across Metazoa
The Pax6 gene and its orthologs control eye development across an extraordinary phylogenetic range, from molluscs and insects to vertebrates [3]. This transcription factor operates as a master regulator of eye development, and its ectopic expression can induce eye formation in unusual body locations. The conservation of Pax6 function across 500 million years of evolution represents a classic example of deep homology, demonstrating that the genetic circuitry for complex organ systems can be maintained over immense evolutionary timescales.
FoxP2 and Vocal Learning Systems
The FoxP2 transcription factor provides a striking example of deep homology extending to neural circuits underlying behavior. FoxP2 plays crucial roles in vocal learning across humans, songbirds, and bats, shaping neural plasticity in cortico-basal ganglia circuits that underlie sensory-guided motor learning [3]. This conservation of genetic regulation for complex behavior demonstrates how deep homology operates beyond morphological structures to include neural systems and cognitive traits.
Table 2: Key Examples of Deep Homology Across Phylogeny
| Genetic Element | Taxonomic Range | Developmental Role | Significance |
|---|---|---|---|
| Pax6 | Mammals, insects, molluscs, cnidarians | Eye development | Master control of eye formation across metazoa |
| Distal-less (Dll/Dlx) | Vertebrates, insects | Limb outgrowth | Patterning of appendages despite independent origins |
| FoxP2 | Humans, songbirds, bats | Vocal learning circuits | Conservation of neural mechanisms for learned behavior |
| Hox genes | Bilaterian animals | Anterior-posterior patterning | Conserved body plan organization across animals |
| Toll-like receptors | Mammals, insects, plants | Innate immunity | Ancient pathogen recognition system |
Methodological Approaches: Experimental and Computational Tools
Experimental Protocols for Deep Homology Research
Spatial Transcriptomics and Single-Cell RNA-Sequencing Protocol
Modern investigations of deep homology employ advanced molecular profiling techniques to map conserved genetic programs. A recent study on the teleost telencephalon exemplifies this approach [4]:
- Tissue Preparation: Dissect telencephala and prepare representative 10μm coronal sections along the rostrocaudal axis
- Spatial Transcriptomics: Capture spatially resolved gene expression profiles using 10x Genomics Visium platform
- Sequence Alignment: Align RNA reads to reference genome (e.g., cichlid Maylandia zebra genome)
- Cell-Type Deconvolution: Map cell populations using algorithms like cell2location to predict anatomical distribution of cell-types identified by snRNA-seq
- Cross-Species Comparison: Compare cell-types and anatomical regions across evolutionary lineages (fish, amphibians, reptiles, birds, mammals)
This integrated approach revealed striking transcriptional similarities between cell-types in the fish telencephalon and subpallial, hippocampal, and cortical cell-types in tetrapods, providing evidence for conserved forebrain organization [4].
Computational Approaches for Detecting Deep Homology
Protein Remote Homology Detection
Advanced computational methods now enable detection of structural and functional homology even when sequence similarity is minimal:
- TM-Vec Framework: A twin neural network model that predicts structural similarity (TM-scores) directly from protein sequences, enabling identification of remote homologs based on structural conservation [5]
- DeepBLAST: A differentiable sequence alignment algorithm that performs structural alignments using protein language models, outperforming traditional sequence alignment methods for remote homology detection [5]
- Dense Homolog Retriever (DHR): An alignment-free method using protein language models and dense retrieval techniques, achieving >10% increase in sensitivity for detecting remote homologs compared to traditional methods [6]
These computational approaches are particularly valuable for annotating proteins of unknown function in metagenomic datasets, where they can identify structural homologs that would be missed by sequence-based methods alone [5].
Table 3: Essential Research Reagents for Investigating Deep Homology
| Reagent/Resource | Type | Function/Application | Example Use |
|---|---|---|---|
| 10x Genomics Visium | Spatial transcriptomics platform | Spatially resolved gene expression profiling | Mapping conserved brain regions across species [4] |
| ProtT5 | Protein language model | Protein sequence embedding and representation | Remote homology detection and structural similarity prediction [7] |
| cell2location | Computational algorithm | Cell-type deconvolution in spatial transcriptomics data | Mapping snRNA-seq cell types to spatial locations [4] |
| TM-align | Structural alignment algorithm | Protein structure comparison and TM-score calculation | Ground truth for training deep learning models [7] |
| CATH Database | Curated protein structure database | Training and benchmarking homology detection methods | Provides structural classifications for model training [7] |
| DHR (Dense Homolog Retriever) | Retrieval framework | Ultra-fast protein homolog detection | Sensitive identification of remote homologs in large databases [6] |
| FoxP2 antibodies | Immunological reagents | Tracking protein expression across species | Comparing neural expression in vocal learning circuits [3] |
Implications and Future Directions
Implications for Biomedical Research
The principles of deep homology have significant implications for drug development and disease modeling. Conserved genetic pathways across species validate the use of model organisms for investigating human disease mechanisms. For example:
- Studies of FoxP2 in songbird vocal learning circuits provide insights into human speech disorders and autism spectrum disorders [3]
- Deep homology in brain organization, as demonstrated by conserved cell-types in the vertebrate forebrain, supports the use of fish models for investigating human neurological and psychiatric conditions [4]
- Conservation of innate immunity pathways (e.g., Toll-like receptors) across animals enables therapeutic development based on model organism studies
Technological Frontiers and Emerging Approaches
Future research in deep homology will be driven by advances in several technological domains:
- Single-cell multi-omics: Simultaneous measurement of gene expression, chromatin accessibility, and protein expression in individual cells across species
- Protein language model advancements: Improved sensitivity for detecting remote homology through models like DHR and Rprot-Vec [6] [7]
- In situ genome editing: CRISPR-based approaches to test functional conservation of regulatory elements in developing embryos across species
- Integration of paleontology and genomics: Combining fossil evidence with molecular data to reconstruct the evolutionary history of genetic toolkits
These approaches will further illuminate how evolution co-opts and modifies conserved genetic toolkits to generate both diversity and novelty in biological systems.
The concept of deep homology has undergone a substantial transformation from Owen's original conception of an abstract archetype to the modern molecular understanding of conserved genetic regulatory networks. This evolutionary developmental framework reveals that despite the remarkable diversity of biological form, a limited set of genetic tools is repeatedly redeployed throughout evolution. For researchers and drug development professionals, this principle provides both practical models for investigating human biology and a profound theoretical framework for understanding the evolutionary constraints and opportunities that shape biological systems. The continuing integration of comparative genomics, single-cell technologies, and computational methods promises to further unravel the deep homologies that underlie biological diversity.
The classical concept of homology, centered on the historical continuity of morphological structures, has been fundamentally transformed by the rise of evolutionary developmental biology (evo-devo). This whitepaper examines how modern frameworks—including gene regulatory network kernels, character identity networks (ChINs), and developmental constraints—provide deeper mechanistic understanding of evolutionary processes. By integrating high-throughput sequencing data and comparative transcriptomics, researchers can now identify deeply conserved genetic circuits that underlie the development of seemingly non-homologous structures across distantly related taxa. These advances reveal that "deep homology" manifests through the conservation of core developmental mechanisms rather than morphological similarity, offering new insights for evolutionary biology and novel approaches for biomedical research.
Homology, originally defined by Sir Richard Owen as "the same organ in different animals under every variety of form and function," has served as a central principle in comparative biology since the pre-Darwinian era [8]. With the advent of evolutionary theory, homology became linked to historical continuity and common descent. However, the distinction between homologous and non-homologous structures has blurred as modern evo-devo has demonstrated that novel features often arise from modification of pre-existing developmental modules rather than emerging completely de novo [8].
The recognition that distantly related species utilize remarkably conserved genetic toolkits during embryogenesis—particularly for patterning fundamental body axes—inspired a reframing of homology with focus on developmental constraints [8]. This conceptual shift led to the formulation of "deep homology," which describes remarkably conserved gene expression during the development of anatomical structures that would not be considered homologous by strict historical definitions [8]. At its core, deep homology helps conceptualize deeper layers of ontogenetic conservation for anatomical features lacking clear phylogenetic continuity.
This whitepaper explores how the integration of next-generation sequencing with conceptual frameworks of kernels, character identity networks, and developmental constraints has revolutionized our understanding of homology in the context of animal design. We examine quantitative evidence, experimental methodologies, and practical research applications that enable researchers to decipher the deep homologies shaping evolutionary trajectories.
Conceptual Frameworks: From Kernels to Character Identity
Gene Regulatory Network Kernels
The kernel concept represents a fundamental principle in the hierarchical organization of gene regulatory networks (GRNs) governing embryogenesis [8]. Kernels constitute sub-units of GRNs that occupy the top of regulatory hierarchies and exhibit specific characteristics:
- Deep evolutionary conservation: Kernels are evolutionarily ancient, often tracing back to phylum or sub-phylum levels
- Functional criticality: They are central to body plan patterning and exhibit resistance to regulatory rewiring
- Stability: Their static nature underlies the remarkable stability observed across animal body plans since the Cambrian explosion
Notable examples of kernel-like GRN conservation include endomesoderm specification in echinoderms, hindbrain regionalization in chordates, and heart development specification in arthropods and chordates [8]. Despite the structural differences between arthropod and chordate hearts, a core set of regulatory interactions directs heart development in both phyla, suggesting a common regulatory blueprint tracing back to a primitive circulatory organ at the base of the Bilateria [8].
Character Identity Networks (ChINs)
Character Identity Networks represent a slightly more flexible approach than kernels for understanding homology [9]. Introduced by Günter Wagner, ChINs refer to the historical continuity of gene regulatory networks that define character identity during development [9]. Unlike kernels, ChINs do not need to be evolutionarily ancient—they can operate at various phylogenetic levels, from phylum down to species.
Central to the ChIN concept is the inherent modularity of developmental systems, where different body parts and organs develop in a semi-autonomous fashion [8]. ChINs underlie this modularity by providing repetitive re-deployment during embryogenesis across generations, while modifications to their output result in varying character states across species [8]. This framework helps resolve conflicts between different lines of evidence, such as embryology versus paleontology, when establishing homologies between morphological characters.
A compelling application of ChIN-based approaches appears in the assessment of digit identity in avian wings. Despite reduction from a pentadactyl ground state to a three-digit formula, comparative RNA-sequencing revealed a strong transcriptional signature uniting the most anterior digits of forelimbs and hindlimbs [8]. This suggests that at the ChIN level, the most anterior digit of the avian wing shares a common developmental blueprint with its hindlimb counterpart, regardless of anatomical position.
Deep Homology and Its Implications
The term "deep homology" was originally coined to describe the repeated use of highly conserved genetic circuits in the development of anatomical features that do not share homology in a strict historical or developmental sense [8]. For example, despite evolutionary separation since the Cambrian and significant morphological divergence, the development of insect and vertebrate appendages shares striking similarities in specifying their embryonic axes [8].
Deep homology extends beyond morphological structures to behavioral traits. Research on FoxP2, a transcription factor relevant for human language, demonstrates its role in shaping neural plasticity in cortico-basal ganglia circuits underlying sensory-guided motor learning across diverse species including humans, mice, and songbirds [3]. This suggests that FoxP2 and its regulatory network may constitute part of a molecular toolkit essential for learned vocal communication, representing a case of deep homology in behavioral systems [3].
Table 1: Key Conceptual Frameworks in Modern Homology Research
| Framework | Key Characteristics | Phylogenetic Scope | Representative Examples |
|---|---|---|---|
| Historical Homology | Based on historical continuity and common descent | All levels | Vertebrate forelimbs; mammalian middle ear bones |
| Kernels | Top-level GRN components; deep conservation; refractory to rewiring | Phylum/sub-phylum level | Heart development (arthropods & chordates); endomesoderm specification |
| Character Identity Networks | Define character identity; developmental modularity; historical continuity | Phylum to species level | Digit identity in avian wings; treehopper helmets |
| Deep Homology | Conserved genetic circuits for non-homologous structures | Distantly related phyla | Appendage development (insects & vertebrates); vocal learning circuits |
Developmental Constraints and Biases
Defining Developmental Constraints
Developmental constraints represent "biases imposed on the distribution of phenotypic variation arising from the structure, character, composition or dynamics of the developmental system" [10]. These constraints collectively restrict the phenotypes that can be produced and influence the directions in which evolutionary change can more easily occur [11]. They can be categorized into three major classes:
- Physical constraints: Limitations imposed by laws of physics (diffusion, hydraulics, physical support) and structural parameters of tissues [11]
- Morphogenetic constraints: Restrictions involving morphogenetic construction rules and self-organizing mechanisms [11]
- Phyletic constraints: Historical restrictions based on the genetics of an organism's development and the need for global induction sequences [11]
A critical reappraisal of developmental constraints argues that the concept should be reframed positively—not as limitations on variation, but as the process determining which directions of morphological variation are possible [10]. From this perspective, development actively "proposes" possible morphological variants in each generation, while natural selection "disposes" of them [10].
The Developmental Hourglass Model
Evidence suggests that constraints are not uniformly distributed throughout development. The earliest developmental stages exhibit remarkable plasticity, while later stages demonstrate extensive diversification [11]. However, during the phylotypic stage—often corresponding to the period of organogenesis—a developmental "bottleneck" occurs where interactions are global and overlapping [11]. This "hourglass" model posits that:
- Early development can accommodate significant changes in morphogen distributions or cleavage planes
- Mid-development features simultaneous, global inductive events that constrain evolutionary change
- Late development consists of compartmentalized, discrete organ-forming systems that can evolve independently
This constrained middle phase of development helps explain why body plans remain stable within phyla despite variations in early and late developmental processes [11].
Table 2: Categories of Developmental Constraints with Examples
| Constraint Type | Basis | Representative Examples |
|---|---|---|
| Physical Constraints | Laws of physics; tissue properties | No vertebrates with wheeled appendages (circulation limitations); size limitations in insects (diffusion constraints) |
| Morphogenetic Constraints | Self-organizing mechanisms; construction rules | Limited digit morphologies in vertebrate limbs (reaction-diffusion mechanisms); forbidden morphologies in salamander limbs |
| Phyletic Constraints | Historical developmental patterns; inductive sequences | Conservation of phylotypic stage across vertebrates; transient notochord requirement in vertebrate embryos |
Quantitative Analysis and Data Visualization
Transcriptomic Evidence for Deep Homology
Next-generation sequencing has revolutionized the detection of deep homology by enabling transcriptome-wide comparisons across species. Spatial transcriptomics and single-nucleus RNA-sequencing provide particularly powerful approaches for identifying conserved cell types and regulatory programs.
Research on the teleost telencephalon demonstrates how these techniques can resolve long-standing questions about evolutionary relationships. Despite the unique "everted" morphology of the teleost telencephalon, comparative analysis of cell-types across fish, amphibians, reptiles, birds, and mammals uncovered striking transcriptional similarities between cell-types in the fish telencephalon and subpallial, hippocampal, and cortical cell-types in tetrapods [4]. This supports partial eversion of the teleost telencephalon and reveals deep homology in vertebrate forebrain organization.
Quantitative analysis of these datasets involves:
- Cross-species alignment of single-cell transcriptomic profiles
- Identification of orthologous cell-type markers
- Spatial mapping of conserved cell populations
- Phylogenetic comparison of gene expression patterns
Comparative Tables of Quantitative Data
Table 3: Representative Evidence for Deep Homology Across Biological Systems
| Biological System | Conserved Elements | Divergent Taxa | Experimental Evidence |
|---|---|---|---|
| Heart Development | NKX2-5/Tinman, TBX5/20, BMP signaling | Arthropods and chordates | Gene expression patterns; knockout phenotypes; regulatory interactions [8] |
| Appendage Patterning | distal-less, homothorax, decapentaplegic/BMP | Insects and vertebrates | Gene expression during limb bud development; functional experiments [8] |
| Vocal Learning Circuits | FoxP2, cortico-basal ganglia circuitry | Humans, songbirds, bats | Gene expression patterns; RNAi knockdown; electrophysiology [3] |
| Forebrain Organization | Pallial, subpallial, hippocampal cell-types | Teleost fish and tetrapods | Single-nucleus RNA-seq; spatial transcriptomics; marker gene analysis [4] |
Experimental Approaches and Methodologies
Transcriptomic Workflows for Deep Homology Detection
The experimental detection of kernels, ChINs, and deep homology relies heavily on modern genomic and transcriptomic approaches. The following diagram illustrates a generalized workflow for identifying deep homology through comparative transcriptomics:
Diagram 1: Experimental workflow for transcriptomic analysis of deep homology
Spatial Transcriptomics for Brain Evolution Studies
A specific application of these methodologies appears in research on vertebrate brain evolution. The following diagram details the integrated approach using single-nucleus RNA-sequencing and spatial transcriptomics to resolve conserved brain cell-types:
Diagram 2: Integrated spatial transcriptomics workflow for brain evolution studies
The Scientist's Toolkit: Research Reagent Solutions
Table 4: Essential Research Reagents and Platforms for Deep Homology Research
| Reagent/Platform | Primary Function | Application Examples |
|---|---|---|
| 10x Genomics Visium | Spatial transcriptomics with morphological context | Mapping cell-type distributions in everted teleost telencephalon; regional annotation of brain areas [4] |
| Single-Nucleus RNA-Seq | High-resolution cell-type classification | Identification of conserved neuronal subtypes across vertebrates; character identity network definition [4] |
| Cell2Location | Bayesian deconvolution of spatial transcriptomics | Mapping snRNA-seq cell-types to spatial coordinates; determining anatomical distributions [4] |
| CRISPR/Cas9 | Gene knockout and genome editing | Functional validation of kernel components; testing necessity of regulatory elements [8] |
| RNAscope/HCR | Multiplexed fluorescence in situ hybridization | Spatial validation of gene expression patterns; co-localization of network components [8] |
| Phylogenetic Footprinting | Comparative genomics for regulatory elements | Identification of conserved non-coding elements; enhancer discovery [8] |
The conceptual transition from historical homology to kernels, character identity networks, and developmental constraints represents a fundamental transformation in evolutionary biology. These frameworks recognize that deep conservation operates primarily at the level of gene regulatory networks and developmental mechanisms rather than morphological structures. The integration of next-generation sequencing technologies with comparative approaches has enabled researchers to decipher these deep homologies across diverse taxa and biological systems.
For researchers and drug development professionals, these advances offer new perspectives on the conservation of biological mechanisms across species. The recognition of deep homology in neural circuits, for example, validates certain animal models for studying human disorders and suggests conserved therapeutic targets. Similarly, understanding developmental constraints provides insight into the permissible versus forbidden morphological variations—knowledge with potential applications in regenerative medicine and tissue engineering.
As single-cell and spatial genomics technologies continue to advance, they will undoubtedly reveal additional examples of deep homology and provide more comprehensive understanding of the kernels and character identity networks that shape evolutionary possibilities. These insights will further bridge the gap between evolutionary theory and biomedical application, demonstrating the enduring utility of homology concepts in contemporary biological research.
The evolution of complex animal body plans is underpinned by a conserved toolkit of intercellular signaling pathways. Among these, Notch, Hedgehog (Hh), and Wnt represent foundational genetic circuits that exhibit remarkable evolutionary conservation from basal metazoans to mammals. These pathways function as central regulators of development, governing processes including cell fate determination, proliferation, and tissue patterning. Recent genomic analyses across diverse taxa have revealed that these signaling systems originated deep in metazoan evolution, with some components predating the emergence of animals altogether. This whitepaper provides an in-depth technical examination of the architecture, evolutionary history, and experimental methodologies for studying these deeply homologous signaling systems, with particular relevance for researchers investigating evolutionary developmental biology and therapeutic target discovery.
The concept of deep homology describes the phenomenon whereby ancient genetic regulatory circuits are redeployed across vast evolutionary distances to build morphologically distinct structures [3]. Notch, Hedgehog, and Wnt signaling pathways exemplify this principle, exhibiting conserved core architectures across the animal phylogeny despite their involvement in the development of divergent anatomical structures.
Molecular analyses reveal that these pathways likely originated before the divergence of major metazoan lineages. Surprisingly, genomic studies of choanoflagellates—the closest living relatives of animals—have identified Notch/Delta pathway components in these unicellular organisms, suggesting that some elements of these signaling systems predate animal multicellularity itself [12]. Similarly, examinations of early-branching metazoans including cnidarians, placozoans, and poriferans have revealed conserved pathway components, shedding light on the ancestral functions of these critical developmental regulators.
The Notch Signaling Pathway
Pathway Architecture and Mechanism
The Notch pathway operates via a relatively simple canonical signaling mechanism that lacks enzymatic amplification steps, making it uniquely sensitive to gene dosage effects [13]. The core signaling mechanism involves proteolytic cleavage of the Notch receptor following ligand binding, leading to translocation of the Notch intracellular domain (NICD) into the nucleus where it regulates transcription of target genes.
Table 1: Core Components of the Notch Signaling Pathway
| Component Type | Mammalian Representatives | D. melanogaster Homologs | Conservation Status |
|---|---|---|---|
| Receptors | NOTCH1, NOTCH2, NOTCH3, NOTCH4 | Notch | Conserved from cnidarians to bilaterians [14] |
| DSL Ligands | DLL1, DLL3, DLL4, JAG1, JAG2 | Delta, Serrate | Broadly conserved; Delta ligands show early diversification [14] |
| Nuclear Effector | RBPJ | Su(H) | Universally conserved |
| Co-activator | MAML | Mastermind | Lost in some lineages including myxozoans [14] |
The ligand-receptor interaction represents a critical regulatory point in Notch signaling. Notch receptors are transmembrane proteins containing multiple epidermal growth factor-like (EGF) repeats in their extracellular domain, while ligands belong to either the Delta-like (DLL) or Jagged (JAG) families [15]. A key regulatory mechanism involves cis-inhibitory interactions, where ligands and receptors expressed on the same cell membrane engage in interactions that render the receptor refractory to trans-activation from neighboring cells [13].
(Diagram 1: Core Notch signaling mechanism)
Evolutionary Conservation Across Metazoa
Comparative genomic analyses of 58 metazoan species reveal broad conservation of core Notch components, with notable losses in certain lineages including ctenophores, placozoans, and some parasitic cnidarians [14]. The canonical Notch pathway likely evolved in the common ancestor of cnidarians and bilaterians, with different lineages exhibiting distinct signaling modes.
Table 2: Notch Pathway Conservation Across Metazoan Lineages
| Lineage | Representative Organisms | Notch Receptor | Ligands | Key Pathway Features |
|---|---|---|---|---|
| Cnidaria | Nematostella vectensis, Hydra vulgaris | Present | Delta, Jagged | Non-canonical (Hes-independent) and canonical signaling modes [14] |
| Porifera | Amphimedon queenslandica | Present | Five Delta ligands | Gene duplications; role in diverse cell types [14] |
| Myxozoa | Sphaerospora molnari | Present | Reduced set | Loss of 14/28 canonical components; extreme genomic reduction [14] |
| Ctenophora | Mnemiopsis leidyi | Present | Absent | Questionable pathway functionality [14] |
In parasitic cnidarians (Myxozoa), extreme genomic reduction has resulted in the loss of approximately 50% of canonical Notch pathway components, including key elements such as MAML, Hes/Hey, and DVL [14]. Despite this reduction, the Notch receptor itself is retained and has been detected in proliferative stages of Sphaerospora molnari, suggesting maintained functionality in cellular proliferation.
Experimental Protocols for Notch Pathway Analysis
Protocol 1: Notch Signaling Inhibition Using Gamma-Secretase Inhibitors
- Compound Preparation: Prepare 100 mM DAPT (N-[N-(3,5-Difluorophenacetyl)-L-alanyl]-S-phenylglycine t-butyl ester) stock solution in DMSO. Aliquot and store at -20°C.
- Treatment Conditions: Apply DAPT at concentrations ranging from 10-100 μM to cultured cells or embryonic specimens. Include DMSO-only controls.
- Exposure Duration: Incubate for 12-48 hours depending on model system and developmental stage.
- Phenotypic Analysis: Assess for Notch-related phenotypes including altered cell differentiation patterns, proliferation defects, or disrupted tissue boundaries.
- Validation: Confirm pathway inhibition through Western blot analysis of NICD levels or qRT-PCR of Notch target genes (Hes/Hey family).
This approach has been successfully applied in diverse systems including cnidarians (Nematostella vectensis, Hydra vulgaris), revealing conserved roles in balancing cell proliferation and differentiation [14].
Protocol 2: Immunohistochemical Localization of Notch Receptors
- Sample Fixation: Fix tissues or whole organisms in 4% paraformaldehyde in PBS for 4-24 hours at 4°C.
- Permeabilization: Treat with 0.1-0.5% Triton X-100 in PBS for 30 minutes to 2 hours.
- Blocking: Incubate in blocking solution (5% normal serum, 1% BSA in PBS) for 1-2 hours.
- Primary Antibody: Apply Notch receptor-specific antibodies at empirically determined dilutions (typically 1:100-1:1000) overnight at 4°C.
- Secondary Detection: Use fluorophore-conjugated secondary antibodies for visualization.
- Counterstaining: Include nuclear counterstains (DAPI, Hoechst) and cytoskeletal markers for context.
This protocol has been adapted for use in non-traditional model systems including myxozoans, demonstrating Notch receptor presence in proliferative cells [14].
The Hedgehog Signaling Pathway
Pathway Architecture and Evolutionary Origin
The Hedgehog (Hh) signaling pathway features a unique mechanism involving autoprocessing of the Hh precursor protein and sterol modification of the active ligand. The Hh protein is synthesized as a precursor that undergoes autocatalytic cleavage to yield an N-terminal signaling domain (hedge) and a C-terminal autoprocessing domain (hog) with intein-like properties [16].
The evolutionary origin of Hh proteins appears to involve domain shuffling early in metazoan evolution. Evidence from sponges and cnidarians reveals the existence of Hedgling—a transmembrane protein containing the Hh N-terminal signaling domain fused to cadherin, EGF, and immunoglobulin domains [17]. This finding suggests that contemporary Hh proteins likely evolved through capture of a hedge-domain by the more ancient hog-domain.
Bacterial homologs of key Hh pathway components provide clues to its deep evolutionary history. Patched (Ptc), the Hh receptor, shows homology to bacterial resistance-nodulation division (RND) transporters [18]. Specifically, a subfamily of RND transporters termed hpnN is associated with hopanoid biosynthesis in bacteria, suggesting an evolutionary connection between sterol transport and Hh signaling.
(Diagram 2: Hedgehog signaling pathway)
Evolutionary Conservation Across Metazoa
Hedgehog signaling components show a complex evolutionary pattern with multiple instances of gene loss and modification. While Drosophila contains a single Hh gene, mammalian genomes possess three paralogs (Shh, Ihh, Dhh) resulting from gene duplication events, with zebrafish exhibiting five Hh genes due to an additional genome duplication in the ray-finned fish lineage [16].
In the nematode Caenorhabditis elegans, a bona-fide Hh gene is absent, replaced by a series of hh-related genes (quahog, warthog, groundhog, and ground-like) that share the Hint/Hog domain but have distinct N-termini [16]. Similar hh-related genes are found in other nematodes including Brugia malayi, suggesting this represents a lineage-specific innovation.
Genomic analyses of the cnidarian Nematostella vectensis reveal six genes with relationship to Hh, including two true Hh genes and additional genes containing Hint/Hog domains with novel N-termini [16]. This diversity suggests that the evolution of hh genes occurred in parallel with the evolution of other Hog domain-containing genes in early metazoan lineages.
Experimental Protocols for Hedgehog Pathway Analysis
Protocol 1: Detection of Hh-Related Genes in Non-Model Organisms
- Sequence Retrieval: Use tblastn and blastp searches with selected Hh, WRT, QUA, GRD, and GRL protein sequences as queries against target genomes or transcriptomes.
- ORF Prediction: Correct predicted ORFs based on conserved domain structure and motif analysis, inspecting genomic sequences for additional exons or alternative splice sites.
- Domain Architecture Analysis: Identify N-terminal signal peptides, Hint/Hog domains, and associated domains using Pfam and SMART databases.
- Phylogenetic Reconstruction: Perform multiple sequence alignments of protein domains followed by phylogenetic analysis using Neighbor Joining and Maximum Likelihood methods.
- Functional Prediction: Analyze sequence motifs within the Hog domain (including motifs J, K, and L) to predict autoprocessing capability and potential cholesterol modification.
This approach has been successfully applied to identify hh and hh-related genes in diverse nematodes and cnidarians [16].
The Wnt Signaling Pathway
Pathway Architecture and Evolutionary Conservation
The Wnt signaling pathway represents one of the most ancient metazoan patterning systems, with evidence of a nearly complete pathway in the simplest free-living animals, placozoans [19]. Sponges, representing one of the earliest branches of metazoa, contain several Wnts and conserved pathway components including Frizzleds, Dickkopf, and Dishevelled [19].
Comparative analyses reveal striking conservation of the chromosomal order of Wnt genes across diverse phyla including cnidarians and bilaterians [19]. The cnidarian Nematostella vectensis possesses an unexpected complexity of Wnt genes, containing almost all subfamilies found in bilaterians, with these genes expressed in patterned domains along the primary body axis during embryonic development [19].
Beta-catenin, the central transcriptional effector of canonical Wnt signaling, shows deeply conserved functions. In sea anemones, beta-catenin is differentially stabilized along the oral-aboral axis, translocates into nuclei at the site of gastrulation, and specifies endoderm, indicating an evolutionarily ancient role in early pattern formation [19].
(Diagram 3: Wnt/β-catenin signaling pathway)
Wnt Pathway Components Across Metazoa
Table 3: Wnt Pathway Conservation Across Metazoan Lineages
| Lineage | Representative Organisms | Wnt Genes | Conserved Components | Functions |
|---|---|---|---|---|
| Placozoa | Trichoplax adhaerens | Present | Complete pathway | Pattern formation |
| Porifera | Sponges | Several Wnts | Frizzled, Dickkopf, Dishevelled | Organizational function [19] |
| Cnidaria | Nematostella vectensis | Most subfamilies | Beta-catenin, TCF | Oral-aboral axis patterning [19] |
| Cnidaria | Hydra | Multiple | Frizzled, beta-catenin, TCF, Dickkopf | Head formation, regeneration |
| Planarians | Girardia tigrina | Present | Conserved pathway | Regeneration polarity [19] |
While Wnt signaling components are conserved throughout animals, some taxa exhibit notable absences. The slime mold Dictyostelium contains Wnt pathway components including a beta-catenin homolog (aardvark) and GSK3, but lacks true Wnt genes themselves [19]. This pattern suggests that the core signaling machinery predates the evolution of the specific Wnt ligands.
Comparative Analysis of Pathway Evolution
The comparative analysis of Notch, Hedgehog, and Wnt signaling pathways reveals both shared and distinct evolutionary patterns. All three pathways originated deep in metazoan history, with some components potentially predating animal multicellularity itself.
Table 4: Comparative Evolutionary Analysis of Signaling Pathways
| Feature | Notch | Hedgehog | Wnt |
|---|---|---|---|
| Earliest Evidence | Choanoflagellates [12] | Cnidarians, sponges [16] [17] | Placozoans, sponges [19] |
| Pre-metazoan Ancestors | Notch/Delta in choanoflagellates [12] | Patched homologs in bacteria [18] | Beta-catenin/GSK3 in slime molds [19] |
| Key Evolutionary Mechanism | Gene duplication in vertebrates | Domain shuffling | Gene family expansion |
| Lineage-Specific Innovations | Cis-inhibitory interactions [13] | hh-related genes in nematodes [16] | Multiple losses in nematodes |
| Developmental Pleiotropy | High (cell fate decisions) [13] | High (patterning, growth) | High (axis patterning) |
A striking pattern across all three pathways is their modular evolution, with components being lost, duplicated, or co-opted in different lineages. For example, while most animals possess a functional Notch pathway, the parasitic cnidarian Myxozoa has lost approximately 50% of its core components [14]. Similarly, nematodes have lost bona-fide Hh genes while evolving novel hh-related genes with distinct N-terminal domains [16].
These pathways also exhibit varying degrees of crosstalk and integration. For instance, the Dishevelled (DVL) protein mediates Wnt-Notch crosstalk, while the gamma-secretase complex cleaves both Notch and other transmembrane proteins including amyloid precursor protein [14]. This molecular crosstalk likely reflects coordinated evolution of these regulatory systems.
The Scientist's Toolkit: Essential Research Reagents
Table 5: Essential Research Reagents for Studying Conserved Signaling Pathways
| Reagent/Category | Specific Examples | Applications | Technical Considerations |
|---|---|---|---|
| Pathway Inhibitors | DAPT (gamma-secretase inhibitor), Cyclopamine (Smo inhibitor), IWP-2 (Wnt inhibitor) | Acute pathway inhibition; functional testing | Dose optimization required; potential off-target effects |
| Genetic Models | Drosophila melanogaster, Caenorhabditis elegans, Nematostella vectensis, Hydra vulgaris | Evolutionary comparisons; functional genetics | Varying genetic tractability; specialized husbandry needs |
| Genomic Resources | Transcriptomes across diverse taxa; genome sequencing databases | Phylogenetic analysis; component identification | Data quality variable; assembly completeness concerns |
| Antibodies | Notch intracellular domain, Patched, Beta-catenin, conserved pathway components | Protein localization; expression analysis | Species cross-reactivity variable; validation required |
| Transgenic Systems | GAL4/UAS (Drosophila), Cre/loxP (mammals), CRISPR/Cas9 systems | Cell-type specific manipulation; gene function analysis | Delivery method optimization; efficiency variation |
The comparative analysis of Notch, Hedgehog, and Wnt signaling pathways reveals the deep evolutionary conservation of developmental genetic circuits across animal phylogeny. These pathways exemplify the principle of deep homology, whereby ancient genetic regulatory circuits are repurposed for novel developmental functions across diverse lineages.
Future research directions should include expanded genomic sampling of early-branching metazoans, particularly understudied lineages such as ctenophores and placozoans, to further resolve the ancestral state of these signaling systems. Functional studies in non-model organisms will be essential for understanding how these conserved pathways have been modified to produce diverse developmental outcomes. Additionally, the exploration of non-canonical signaling modes and pathway crosstalk in basal metazoans may reveal ancestral functions that have been obscured in more derived model systems.
From a therapeutic perspective, the deep conservation of these pathways underscores their fundamental importance in cellular regulation while also highlighting potential challenges for targeted interventions due to pleiotropic effects. Understanding the evolutionary context of pathway modifications may inform the development of more specific therapeutic approaches that target lineage-specific innovations while sparing conserved core functions.
The independent evolution of complex anatomical structures in distantly related species, such as limbs in insects and vertebrates or hearts in arthropods and chordates, has long intrigued evolutionary and developmental biologists. The concept of deep homology provides a powerful explanatory framework for these phenomena. Deep homology refers to the sharing of ancestral genetic regulatory circuits that are used to build morphologically and phylogenetically disparate structures [20]. This principle posits that new anatomical features do not typically arise de novo but rather evolve from pre-existing genetic regulatory networks established early in metazoan evolution [20] [21]. These conserved developmental kernels provide a shared toolkit that can be co-opted, modified, and elaborated upon in different lineages to generate evolutionary novelty.
This whitepaper examines two paradigmatic case studies through the lens of deep homology: limb development and heart specification. The analysis reveals that despite vast phylogenetic distances and fundamentally different anatomical organizations, insects and vertebrates utilize conserved molecular machinery for patterning their appendages. Similarly, the genetic programs underlying heart development in arthropods and chordates share common evolutionary origins. For research scientists and drug development professionals, understanding these deeply conserved mechanisms provides valuable insights into congenital disorders and reveals potential therapeutic targets that operate across multiple tissue types and organ systems.
Case Study 1: Limb Development from Insects to Vertebrates
Conserved Genetic Circuitry in Limb Patterning
Limb development proceeds through four principal phases: (1) initiation of the limb bud, (2) specification of limb pattern, (3) differentiation of tissues, and (4) shaping of the limb and its growth to adult size [22]. Remarkably, the core genetic pathways governing these processes exhibit profound conservation between insects and vertebrates, representing a classic example of deep homology.
The limb system serves as a model for pattern formation within the vertebrate body plan, with the same molecular toolkits deployed at different times and places in vertebrate embryos [22]. Genetic studies have revealed that the Hox gene family, which specifies positional identity along the anterior-posterior axis, is utilized in patterning both insect legs and vertebrate limbs. Similarly, the Distal-less (Dll) gene, first identified for its role in distal limb development in Drosophila, plays a conserved role in specifying distal structures in vertebrate appendages. The Notch signaling pathway, which regulates cell fate decisions through local cell interactions, is another deeply conserved component that patterns the joints of both arthropod and vertebrate limbs [21].
The conservation extends beyond single genes to encompass entire regulatory circuits. As Shubin and colleagues noted, animal limbs of every kind—from whale flippers and fish fins to bat wings and human arms—are "organized by a similar genetic regulatory system that may have been established in a common ancestor" [21]. This shared genetic architecture facilitates the independent evolution of diverse limb morphologies through modifications to the regulation, timing, and combinatorial use of a common developmental toolkit.
Evolutionary Dynamics and Limb Evolvability
While limbs are serially homologous structures that share a common genetic architecture, they can evolve independently when selective pressures differ between forelimbs and hindlimbs. This evolutionary independence is particularly evident in humans, whose distinctive limb proportions (long legs and short arms) represent adaptations for bipedalism [23].
Quantitative analyses of limb integration in anthropoid primates reveal how developmental constraints have been modified throughout evolution. Humans and apes exhibit significantly reduced integration between limbs (34-38% reduced) compared to quadrupedal monkeys, enabling greater independent evolvability of limb proportions [23]. This reduction in integration reflects alterations to the pleiotropic effects of genes that normally constrain limb development, allowing for the mosaic pattern of evolution observed in the hominin fossil record.
Table 1: Limb Integration Patterns in Anthropoid Primates
| Species Category | Limb Integration Strength | Homologous Element Correlation | Evolutionary Disparity |
|---|---|---|---|
| Quadrupedal Monkeys | High | Strong (Fisher-z: 1.22-1.50) | Lower |
| Apes | Reduced (34-38% less than monkeys) | Moderate (Fisher-z: 1.00-1.08) | Intermediate |
| Humans | Reduced (34-38% less than monkeys) | Moderate (Fisher-z: 0.93) | Higher |
This evolutionary perspective has practical implications for biomedical research. The modular nature of limb development means that genetic variants or chemical perturbations can affect different limbs differently. Understanding the mechanisms that both integrate and dissociate limb development provides insights into congenital conditions that affect specific appendages while sparing others.
Experimental Approaches for Limb Development Research
The study of limb development employs sophisticated molecular, genetic, and genomic techniques. Below are key methodological approaches for investigating the deep homology of limb patterning mechanisms.
Table 2: Key Research Reagents for Limb Development Studies
| Research Reagent | Application in Limb Development | Example Use Case |
|---|---|---|
| CRISPR/Cas9 mutagenesis | Gene knockout and functional analysis | Tissue-specific mutagenesis of Pax3/7 in neuronal development [24] |
| Electroporation | Introduction of plasmids into specific tissues | Ectopic overexpression of transcription factors [24] |
| Cis-regulatory reporter analysis | Identification of regulatory elements | Studying transcriptional regulation in neuronal networks [24] |
| Lineage tracing (e.g., Cre-Lox) | Cell fate mapping and lineage analysis | Tracing neural crest contributions [25] |
| Transcriptome analysis | Gene expression profiling | Microarray-based transcriptome of isolated neurons [24] |
Limb Patterning Conservation Across Species
Protocol: CRISPR/Cas9 Mutagenesis for Functional Gene Analysis
- sgRNA Design: Identify target sequences in genes of interest (e.g., Pax3/7) using specialized software [24].
- Electroporation: Introduce CRISPR/Cas9 constructs into dechorionated fertilized zygotes using square-wave electroporation (e.g., 6-12V, 5-10 ms pulse length) [24].
- Screening: Assess mutagenesis efficiency via PCR and sequencing of target loci.
- Phenotypic Analysis: Examine limb phenotypes using morphological assessment and molecular markers.
- Validation: Confirm specificity through rescue experiments and off-target effect assessment.
This approach enables researchers to functionally test the role of deeply conserved genes in limb development and assess whether their functions are maintained across evolutionary distant species.
Case Study 2: Heart Specification in Arthropods and Chordates
Evolutionary Origins of Cardiac Structures
The cardiovascular system has undergone substantial evolutionary modification from its origins in primitive contractile cells to the complex multi-chambered hearts of birds and mammals. The earliest contractile proteins appeared approximately 2 billion years ago during the Paleoproterozoic Era, with contractile cells eventually organizing into primitive tubes that moved fluid via peristaltic-like contractions [26]. This primitive tubular pumping structure in chordates represents the evolutionary blueprint for cardiac circulatory systems in both invertebrates and vertebrates through conserved homologies [26].
In the transition from water to land, vertebrates evolved increasingly complex cardiac structures to support higher metabolic demands. Teleost fish possess a four-chambered heart in series (sinus venosus, atrium, ventricle, and bulbus arteriosus) that supports a single circulation system [26]. With the emergence of air-breathing vertebrates, circulatory systems separated into pulmonary and systemic circuits, culminating in the fully septated, four-chambered hearts of birds and mammals that allow complete separation of oxygenated and deoxygenated blood [26].
Table 3: Evolutionary progression of heart structures across vertebrates
| Species Group | Heart Structure | Circulation Type | Key Features |
|---|---|---|---|
| Teleost Fish | Four chambers in series | Single circulation | Sinus venosus, atrium, ventricle, bulbus arteriosus |
| Non-crocodilian Reptiles | Two atria, one partially divided ventricle | Partial separation | Blood mixing capability; shunting ability |
| Crocodilians | Two atria, two ventricles | Dual circulation with shunting | Two aortic outlets; diving adaptations |
| Birds and Mammals | Two atria, two ventricles | Complete dual circulation | Full septation; high-pressure systemic circuit |
Despite this structural diversity, the genetic and developmental foundations of heart specification reveal deep homologies between arthropods and chordates. The Tinman/Nkx2-5 gene family, first identified for its essential role in Drosophila heart development, has orthologs that play conserved roles in vertebrate cardiogenesis. Similarly, core signaling pathways including BMP, Wnt, and Notch regulate heart development across bilaterians, reflecting their ancestral roles in patterning the contractile vasculature.
Gene Regulatory Networks in Heart Development
Cardiac development is governed by evolutionarily conserved gene regulatory networks (GRNs) that exhibit modular organization. Studies in tunicates (Ciona robusta), as the sister group to vertebrates, have revealed conserved GRNs for specifying particular cardiac cell types [24]. The combinatorial and modular logic of these networks allows for a diversity of cardiac morphologies through the redeployment of conserved regulatory modules.
The GRN controlling the specification of putative Mauthner cell homologs in tunicates illustrates this modular principle. The transcription factor Pax3/7 sits atop a regulatory hierarchy that controls neuronal specification and differentiation, operating through downstream factors including Pou4, Lhx1/5, and Dmbx that regulate distinct branches of the network dedicated to different developmental tasks [24]. Homologs of these transcription factors are similarly essential for cranial neural crest specification in vertebrates, indicating deep conservation of this regulatory circuitry [24].
The modular organization of cardiac GRNs has important implications for evolutionary innovation and medical genetics. Mutations in deeply conserved components often cause severe congenital heart defects, while modifications to regulatory linkages between modules can drive evolutionary changes in heart structure without compromising core cardiac functions.
Neural Crest Contributions and Cardiac Evolution
A pivotal innovation in vertebrate heart evolution was the contribution of the cardiac neural crest, an ectodermal cell population that migrates into the pharyngeal arches and contributes to the aortic arch arteries and arterial pole of the heart [25]. First demonstrated in avian embryos through neural crest ablation experiments, this cell population gives rise to the smooth muscle of the great arteries and plays essential roles in outflow tract septation and arch artery remodeling [25].
The molecular regulation of cardiac neural crest development involves a conserved genetic program including Tbx1, haploinsufficiency of which causes DiGeorge syndrome (22q11.2 deletion syndrome) with characteristic cardiovascular malformations [25]. The deep homology of this genetic program is evident from its conservation across vertebrates and its relationship to more primitive cell migration programs in invertebrate chordates.
Heart Development Evolutionary Pathway
Protocol: Neural Crest Lineage Tracing and Ablation
- Lineage Labeling: Use Cre-Lox technology (e.g., Wnt1-Cre or Pax3-Cre mice) to lineage label neural crest cells [25].
- Ablation Studies: Perform bilateral ablation of neural crest populations (e.g., over somites 1-3 in avian embryos) [25].
- Chimera Generation: Create quail-chick chimeras by grafting quail neural crest into host chick embryos [25].
- Phenotypic Analysis: Assess cardiovascular defects, particularly outflow tract and aortic arch malformations.
- Molecular Characterization: Analyze expression of key regulators (e.g., Tbx1) in manipulated embryos.
These experimental approaches have been instrumental in elucidating the essential contributions of neural crest cells to cardiovascular development and the deep homology of the genetic programs guiding their development.
Research Applications and Future Directions
The Scientist's Toolkit: Core Reagents and Technologies
Contemporary research into deep homology leverages an expanding toolkit of molecular, genomic, and computational technologies. The ongoing technological revolution in developmental biology is accelerating progress through advances in genomics, imaging, engineering, and computational biology [27].
Table 4: Essential research reagents for evolutionary developmental biology
| Technology/Reagent | Application | Utility for Deep Homology Studies |
|---|---|---|
| Single-cell RNA sequencing | Transcriptome profiling | Identifying conserved cell types and states across species |
| CRISPR/Cas9 genome editing | Gene functional analysis | Testing necessity and sufficiency of conserved genes |
| Live imaging and light-sheet microscopy | Dynamic morphogenesis | Comparing developmental processes across species |
| Organoid systems | In vitro modeling | Reconstituting conserved developmental programs |
| Cross-species chromatin profiling | Regulatory element identification | Discovering deeply conserved enhancers |
Implications for Biomedical Research and Therapeutic Development
Understanding deep homology has profound implications for biomedical research and therapeutic development. The conservation of genetic programs across diverse species means that mechanistic insights gained from model organisms often translate to human biology and disease. Furthermore, the modular nature of developmental gene regulatory networks suggests that therapeutic interventions could be designed to target specific network modules without disrupting entire systems.
For drug development professionals, the deep homology concept provides a framework for prioritizing targets with evolutionarily conserved functions, which may offer broader therapeutic windows and fewer off-target effects. Additionally, understanding how developmental processes are conserved enables more predictive toxicology assessments during preclinical development.
The entrance of developmental biology into "a new golden age" driven by powerful technologies [27] promises to further illuminate the deep homologies underlying animal development. As these advances continue, they will undoubtedly reveal new opportunities for therapeutic intervention in congenital disorders and regenerative medicine approaches for damaged tissues and organs.
The concept of homology represents a foundational pillar in comparative biology, representing the relationship among characters due to common descent. Within the context of animal design research, homology operates across multiple hierarchical levels—from molecular and cellular to morphological and developmental—creating a complex framework of "sameness" that illuminates evolutionary relationships. Historically, homology was defined morphologically and explained by reference to ideal archetypes, implying design. Charles Darwin reformulated biology in naturalistic terms, explaining homology as the result of descent with modification from a common ancestor. This phylogenetic definition has since dominated evolutionary biology, though the fundamental challenge remains: how to objectively identify and validate homologies across deep evolutionary divergences where structural similarities become obscured by eons of evolutionary change [28].
The emerging field of evolutionary developmental biology (evo-devo) has revealed that hierarchical homology operates through deeply conserved genetic and developmental pathways, often called "deep homology," where analogous structures in distantly related species share common genetic regulatory apparatus. This whitepaper synthesizes current research and methodologies for identifying homology across biological hierarchies, with particular emphasis on applications in biomedical research and drug discovery. By integrating classical morphological approaches with cutting-edge genomic technologies, researchers can now trace homological relationships across vast evolutionary distances, providing unprecedented insights into animal design principles with practical applications in human health and disease modeling.
Theoretical Foundations: From Morphology to Molecular Regulation
Historical Perspectives and Definitions
The historical development of homology concepts reveals shifting explanatory frameworks. Pre-Darwinian biologists like Richard Owen defined homology strictly morphologically as "the same organ in different animals under every variety of form and function." Darwin's revolutionary contribution was to provide a naturalistic mechanism—descent with modification—to explain these similarities. This subsequently led to a redefinition of homology in phylogenetic terms as features derived from the same feature in a common ancestor [28]. This phylogenetic definition creates a logical circularity if used to prove common ancestry, highlighting the need for independent criteria for establishing homology.
The hierarchical nature of homology becomes apparent when considering the complex relationship between genetic, developmental, and morphological levels. As noted by evolutionary biologist Leigh Van Valen, homologous features are produced during development by information that has been inherited with modification from ancestors, creating a "continuity of information" across generations [28]. This informational perspective bridges the gap between phylogenetic patterns and developmental processes, allowing homology to be traced through inherited developmental programs despite morphological diversification.
The Challenge of Deep Homology
A significant challenge in evolutionary biology involves establishing homology across deep evolutionary divergences where morphological similarities are obscured. The central problem is that high genomic evolvability and the complexity of genomic features that impact gene regulatory networks make it difficult to identify clear shared molecular signatures for homologous cell types or structures between deeply branching animal clades [29]. Complex interplay between regulatory networks during development and the transcription factor logic associated with cell types makes identifying a clear shared set of genes that identify a given cell type challenging for clades separated for hundreds of millions of years.
Table 1: Levels of Hierarchical Homology with Defining Characteristics
| Level | Defining Characteristics | Evolutionary Lability | Evidence Methods |
|---|---|---|---|
| Genetic | Similar DNA sequences, syntenic relationships | Low (sequence conservation) | Genome sequencing, alignment algorithms [30] |
| Genomic Architecture | Irreversible chromosomal mixing, regulatory entanglements | Very Low (synapomorphic) | Chromosomal-scale genomes, Hi-C, synteny analysis [29] |
| Developmental | Conserved gene regulatory networks, cell lineage | Moderate (developmental system drift) | Single-cell transcriptomics, lineage tracing, CRISPR [29] |
| Cellular | Molecular signatures, ultrastructural features | Moderate-High | ImmunoFISH, proteomics, electron microscopy [31] |
| Morphological | Anatomical position, topological relationships | High (adaptive convergence) | Comparative anatomy, fossil evidence, 3D reconstruction [28] |
Genomic Approaches: Irreversible States as Homology Markers
Irreversible Genomic States as Synapomorphic Characters
A groundbreaking approach to establishing deep homologies leverages the concept of irreversible genomic states. These states occur after chromosomal and sub-chromosomal mixing of genes and regulatory elements, creating configurations that cannot revert to ancestral conditions. Similar to historical definitions of homology based on anatomical unmovable components, these genomic states provide stable reference points for tracing evolutionary relationships [29]. The key insight is that while many genomic changes can be reversed through evolution, certain configurations—particularly those involving complex rearrangements—effectively "lock in" evolutionary histories.
The most characterized form of irreversible genomic change is "fusion-with-mixing"—when two ancestrally conserved chromosomes undergo fusion, followed by intra-chromosomal translocations that mix genes from both original chromosomes. The resulting mixed chromosome cannot be reverted to the original two states comprising the two ancestral gene complements [29]. This chromosomal-scale mixing creates a powerful synapomorphic character that, once established, cannot be reverted and is expected in all descendants of that lineage. This property has been utilized to resolve previously debated phylogenetic positions where morphological or sequence-based approaches yielded conflicting results.
Sub-Chromosomal Regulatory Entanglements
Beyond chromosomal fusions, a parallel process occurs at the sub-chromosomal level through what has been termed "regulatory entanglement." Mixing of enhancer-promoter interactions within topologically associating domains (TADs) or loop structures may create configurations unlikely to be unmixed by random inversions, as these would break functional enhancer-promoter contacts [29]. These constrained genomic neighborhoods result in the retention of unrelated genes and their regulatory regions, creating irreversible genomic states that can be linked to specific cell type identities or developmental processes.
The irreversibility of this evolutionary process enables researchers to screen such states for specific changes in gene expression associated with cell type development or function. Phylogenetic dating of such regulatory entanglements and quantification of their irreversibility can indicate at what evolutionary node a novelty arose and rule out scenarios of re-ancestralization. The methodology for identifying such states is emerging, building on novel interdisciplinary applications including topological theories in macro-evolution [29]. This approach provides a fertile testing ground for deep evolutionary phenotype homology hypotheses that were previously intractable.
Experimental Methodologies for Establishing Homology
Chromosomal Organization Analysis
The investigation of homologous chromosome pairing provides a powerful experimental model for studying genomic organization. In one representative study, researchers employed immunofluorescence and DNA fluorescence in situ hybridization (ImmunoFISH) with high-resolution confocal microscopy to visualize chromosomes and centrosomes in human endothelial cells [31]. The experimental workflow followed this detailed protocol:
Cell Culture and Preparation: Primary human umbilical vein endothelial cells (HUVECs) and human aortic endothelial cells (HAECs) from individual donors were cultured on flamed/UV-sterilized PTFE glass slides until reaching 70-80% confluency. The specific culture medium consisted of MCDB-131 supplemented with 1% Glutamax, 1% Pen-Strep, and 2% large vessel endothelial supplement [31].
Immunofluorescence Protocol: Slides were fixed with 4% paraformaldehyde and stored in 70% ethanol for at least 24 hours. After washing in cold PBS, heat-induced antigen retrieval was performed for 10 minutes in sodium citrate buffer (10 mM sodium citrate, 0.05% Tween-20, pH 6.0) in a steamer. Slides were permeabilized (0.25% Triton X-100 in PBS), blocked with 10% goat serum, and incubated with primary antibody against γ-tubulin (1:1000 dilution) at 4°C overnight in a humidified chamber. Following PBS washes, slides were incubated with Alexa Fluor 647-conjugated secondary antibody (1:500) for 1 hour at room temperature in darkness [31].
Chromosome Painting: Before DNA counterstaining, slides were incubated with EGS crosslinker solution (25% DMSO, 0.375% Tween-20, 25mM EGS in PBS) for 10 minutes. Whole Chromosome Paints for specific chromosomes in Aqua, Texas red, or FITC were preheated at 80°C for 10 minutes, then incubated at 37°C for 1 hour [31].
Imaging and Analysis: High-resolution confocal microscopy enabled 3D reconstruction of chromosome positions relative to centrosomes. This allowed quantification of homologous chromosome pairing frequencies by determining whether homologs resided on the same or opposite sides of the centrosome axis [31].
Table 2: Research Reagent Solutions for Chromosomal Homology Studies
| Reagent/Tool | Specifications | Function in Experiment |
|---|---|---|
| HAECs | Lonza Cat. No. CC-2535 (multiple donor ages) | Provides adult endothelial cells for age-related comparison studies [31] |
| γ-tubulin Antibody | abcam ab11317 (Rabbit, 1:1000 dilution) | Labels centrosomes for spatial reference in chromosome positioning [31] |
| Alexa Fluor 647 Secondary | abcam ab150079 (Goat Anti-Rabbit, 1:500) | Fluorescent detection of primary antibody for confocal imaging [31] |
| Whole Chromosome Paints | Applied Spectral Imaging (Aqua, Texas red, FITC) | Fluorescently labels specific chromosomes for visualization [31] |
| EGS Crosslinker | Thermo Scientific #21565 (25mM in PBS) | Crosslinks proteins to maintain structural integrity during FISH [31] |
Homology Modeling in Structural Biology
Homology modeling represents a computational approach to establish structural homology when experimental structures are unavailable. The process involves predicting a protein's 3D structure based on its alignment to related proteins with known structures. This method relies on the principle that structural conformation is more conserved than amino acid sequence, and small-to-medium sequence changes typically result in minimal 3D structure variation [30]. The homology modeling process consists of five key steps: (1) template identification through fold recognition; (2) single or multiple sequence alignment; (3) model building based on template 3D structure; (4) model refinement; and (5) model validation [30].
Template Recognition and Alignment: The initial step uses tools like BLAST to compare the target sequence against the Protein Data Bank (PDB). For sequences with identity below 30%, more sensitive methods like PSI-BLAST, Hidden Markov Models (HMMER, SAM), or profile-profile alignment (FFAS03) are required. Alignment accuracy is critical, as errors become the primary source of deviations in comparative modeling [30].
Model Building and Refinement: After target-template alignment, model building employs methods including rigid-body assembly, segment matching, spatial restraint satisfaction, and artificial evolution. Model refinement uses energy minimization with molecular mechanics force fields, complemented by molecular dynamics, Monte Carlo, or genetic algorithm-based sampling [30]. The accuracy of the resulting model directly correlates with sequence identity between target and template—models with >50% identity are generally accurate enough for drug discovery applications, while those with 25-50% identity may guide mutagenesis experiments [30].
Quantitative Frameworks and Data Visualization
Standards for Homology Assessment
Establishing quantitative thresholds is essential for robust homology assessment across biological hierarchies. The criteria vary significantly depending on the hierarchical level being investigated, with more stringent requirements at molecular levels where homoplasy (convergent evolution) is less likely. Current frameworks distinguish between different strengths of evidence, with genomic architectural features providing the strongest evidence due to their irreversible nature [29].
Table 3: Quantitative Thresholds for Homology Assessment Across Hierarchies
| Homology Level | Strong Evidence Threshold | Moderate Evidence | Key Metrics |
|---|---|---|---|
| Sequence Homology | >50% identity (structural) >70% (functional) | 25-50% identity | E-value, Bit score, Alignment coverage [30] |
| Structural Homology | RMSD <2Å, TM-score >0.5 | RMSD 2-4Å, TM-score 0.3-0.5 | Root-mean-square deviation, Template Modeling score [30] |
| Gene Expression | >0.85 correlation coefficient | 0.7-0.85 correlation | Pearson's r, Spearman's ρ, Jaccard index [32] |
| Chromosomal Pairing | >70% antipairing frequency | 50-70% antipairing | Distance to centrosome, homologous separation [31] |
| Genomic Entanglement | Irreversible state + functional link | Irreversible state only | Synteny conservation, TAD boundary conservation [29] |
Data Visualization for Homology Relationships
Effective visualization is crucial for interpreting complex hierarchical homology relationships. Different visualization methods serve distinct purposes in homology research, from comparing genetic distances to representing structural alignments and genomic architectures. The choice of visualization method should be guided by the specific research question and the nature of the homology being investigated [32].
Phylogenetic trees with evolutionary distance metrics represent one of the most established visualization methods in homology studies. These trees depict evolutionary relationships between species or genes, with branch lengths proportional to genetic distance (measured as mutations or changes between sequences). Color coding can represent evolutionary time or mutation rates, helping biologists understand how different taxa evolved from common ancestors and providing insight into speciation events and evolutionary timescales [32].
For structural homology, 3D molecular models show protein folding and structure, with different regions color-coded based on stability, function, or molecular interactions. Quantitative data such as bond length or hydrophobicity can be overlaid on the structure to highlight functional regions, enabling biochemists to study how proteins fold and interact with other molecules—information essential for drug design and understanding disease mechanisms [32].
Applications in Drug Discovery and Biomedical Research
Structure-Based Drug Design
Homology modeling has become an indispensable tool in structure-based drug discovery, particularly for target classes with limited experimental structural data. Recent advances demonstrate that high-quality predicted structures enable structure-based approaches for an expanding number of drug discovery programs. When combined with free energy perturbation (FEP) calculations, predicted structures can be confidently employed to achieve drug design goals, even for challenging targets like the hERG potassium channel [33]. This approach is particularly valuable for membrane proteins and other target classes that are difficult to characterize experimentally.
The application of homology modeling in drug discovery spans multiple stages, from target identification and validation to lead optimization. By providing 3D structural information for proteins that would otherwise be inaccessible, homology modeling expands the druggable genome and enables structure-based approaches for target classes previously limited to ligand-based methods. The reliability of these models has improved significantly through advances in template recognition, alignment algorithms, and model refinement techniques, making them valuable tools for rational drug design [30].
Disease Modeling through Evolutionary Homologies
Understanding hierarchical homology enables more effective disease modeling by leveraging evolutionary relationships between model organisms and humans. The conservation of gene regulatory networks, cell type identities, and developmental processes across species provides the foundation for using model organisms to study human disease mechanisms. Recent approaches using irreversible genomic states to define and trace ancient cell type homologies offer new opportunities for understanding disease susceptibility and pathogenesis from an evolutionary perspective [29].
The finding that homologous chromosome pairing frequencies change with cellular age illustrates how basic homology relationships can inform disease mechanisms. Research demonstrates that mitotic antipairing is lost in adult aortic endothelial cells, with small chromosomes showing more frequent pairing abnormalities. This age-dependent loss of chromosomal spatial organization may contribute to increased genomic instability and disease susceptibility in aging tissues, particularly in the cardiovascular system [31]. Such insights highlight how fundamental research into homology mechanisms can illuminate pathological processes.
Future Directions and Concluding Perspectives
The field of hierarchical homology is advancing rapidly through integration of multi-omics data, computational modeling, and high-resolution imaging. Emerging approaches focus on identifying irreversible genomic states as stable reference points for establishing deep homologies across evolutionary timescales. These methods leverage chromosomal and sub-chromosomal mixing events that create essentially permanent genomic signatures of evolutionary history [29]. As these approaches mature, they will enable more robust reconstruction of ancestral states and more accurate dating of evolutionary innovations.
The practical applications of hierarchical homology in drug discovery and biomedical research continue to expand. Structure-based drug design utilizing predicted models is becoming increasingly routine, while understanding the evolutionary relationships between model organisms and humans improves preclinical research translation. Future advances will likely come from integrating hierarchical homology data across biological scales—from genomic rearrangements to protein structures—to create unified models of biological systems that bridge evolutionary and biomedical sciences. This integration will further establish hierarchical homology as a foundational concept for understanding animal design and applying this understanding to address human health challenges.
From Sequence to Function: Cutting-Edge Methods for Detecting and Applying Deep Homology
The integration of Next-Generation Sequencing (NGS) and transcriptomics has fundamentally transformed homology detection from a gene-by-gene analysis to a comprehensive, genome-wide investigation. This paradigm shift is pivotal for research in deep homology, the principle that conserved genetic regulatory circuits underlie the development of anatomically diverse structures across the animal kingdom. This technical guide elucidates how NGS technologies, coupled with advanced bioinformatic tools, enable the uncovering of these ancient evolutionary relationships by providing unprecedented access to genomic and transcriptomic data on a massive scale. We detail experimental protocols, data analysis workflows, and key reagent solutions that empower researchers to probe the molecular foundations of animal design.
Deep homology refers to the existence of shared, ancient genetic mechanisms for building morphologically and phylogenetically disparate biological structures. Uncovering these relationships requires the ability to compare genetic regulatory networks across a wide range of species, a task that was historically limited by technological constraints.
The advent of Next-Generation Sequencing (NGS) has broken these barriers. NGS is a massively parallel sequencing technology that provides ultra-high throughput, scalability, and speed, allowing for the determination of nucleotide order in entire genomes or targeted regions of DNA or RNA [34]. The application of NGS to transcriptomics—known as RNA-Seq—enables researchers to discover novel RNA variants, quantify mRNAs for gene expression analysis, and comprehensively profile the transcriptome [34]. By generating millions of sequences from a single sample, NGS provides the necessary data density to move beyond the detection of isolated homologous genes to the system-level identification of homologous networks and pathways that constitute the deep logical structure of animal development.
NGS Technology: Foundational Platforms and Principles
NGS encompasses several platforms, each with distinct advantages tailored to different aspects of genomic and transcriptomic analysis. The core workflow involves library preparation, sequencing, and data analysis [34].
Core Sequencing Technologies
Table 1: Comparison of Key Next-Generation Sequencing Platforms.
| Platform | Sequencing Technology | Read Length | Key Advantages | Primary Applications in Homology |
|---|---|---|---|---|
| Illumina [35] [34] | Sequencing-by-Synthesis (SBS) | Short (36-300 bp) | High throughput, low error rates, cost-effective | Whole-genome sequencing, RNA-Seq, variant detection, high-coverage applications |
| PacBio SMRT [35] [36] | Single-Molecule Real-Time | Long (avg. 10,000-25,000 bp) | Long reads, detects epigenetic modifications | Resolving complex genomic regions, structural variation, full-length transcript sequencing |
| Oxford Nanopore [35] [37] | Electrical Impedance Detection | Long (avg. 10,000-30,000 bp) | Real-time sequencing, portability, direct RNA sequencing | Rapid sequencing, field applications, identifying splice variants |
| Ion Torrent [35] [37] | Semiconductor Sequencing | Short (200-400 bp) | Rapid turnaround, simple workflow | Targeted sequencing, small-genome sequencing |
Targeted Sequencing Approaches
For focused studies on specific gene families or regulatory regions, targeted sequencing offers a cost-effective strategy. Exome sequencing, which focuses on the protein-coding regions of the genome (1-2% of the total genome), is a powerful example [38]. It allows for the deep sequencing of functionally relevant genetic variations across numerous samples. Similarly, targeted sequencing using long-read technologies like PacBio's SMRT sequencing enables researchers to obtain haplotype-resolved data and resolve difficult-to-sequence regions, which is crucial for accurately characterizing complex gene families [36].
From Sequence to Homology: Theoretical and Practical Foundations
Inferring Homology from Sequence Similarity
Homology, defined as common evolutionary ancestry, is inferred from statistically significant sequence similarity [39]. The underlying principle is that when two sequences share more similarity than would be expected by chance, the simplest explanation is that they arose from a common ancestor. Statistical tools like BLAST, FASTA, and HMMER are used to calculate the significance of an alignment, typically reporting an E-value, which estimates the expected number of times a score would occur by chance in a database of a given size [39].
It is critical to distinguish between protein-based and DNA-based searches. Protein sequence comparisons are vastly more sensitive for detecting remote homology because the evolutionary "look-back time" is 5 to 10 times longer than for DNA-DNA comparisons. Protein-protein alignments can routinely detect homology in sequences that diverged over 2.5 billion years ago, whereas DNA-DNA alignments rarely detect homology beyond 200-400 million years of divergence [39].
The Shift to Alignment-Free and Genome-Wide Methods
Traditional methods rely on sequence alignment, which can be computationally intensive and may miss highly divergent homologs. Recent innovations, such as the Dense Homolog Retriever (DHR), leverage protein language models, dense retrieval, and contrastive learning to detect homology in an alignment-free manner [40]. This approach is particularly adept at unveiling subtle homology relationships and offers remarkable acceleration, sometimes by orders of magnitude, making large-scale database searches to explore protein sequence diversity feasible [40].
Experimental Protocols for Genome-Wide Homology Detection
This section provides detailed methodologies for key experiments that leverage NGS and transcriptomics to identify deep homologs.
Protocol 1: Comparative Transcriptomics for Gene Expression Profiling
Objective: To identify conserved gene expression patterns across different species or tissues during development.
- Sample Collection & RNA Extraction: Collect tissue samples from relevant developmental stages across multiple species. Preserve samples immediately in RNAlater. Extract total RNA using a kit-based method (e.g., TRIzol), ensuring RNA Integrity Number (RIN) > 8.0.
- Library Preparation: Use a stranded mRNA-Seq library prep kit. Enrich poly-A containing mRNA using oligo-dT beads. Fragment purified mRNA and synthesize cDNA. Ligate sequencing adapters and perform PCR amplification to enrich for adapter-ligated fragments.
- Sequencing: Pool libraries and sequence on an Illumina platform (e.g., NovaSeq) to generate a minimum of 30 million paired-end (e.g., 2x150 bp) reads per sample.
- Data Analysis:
- Quality Control: Use FastQC to assess read quality. Trim adapters and low-quality bases with Trimmomatic.
- Alignment & Quantification: Map reads to the respective reference genomes of each species using a splice-aware aligner like STAR. Quantify reads per gene feature using featureCounts.
- Orthology Mapping: Use OrthoFinder to identify orthologous gene groups (orthogroups) across all species in the study.
- Expression Analysis: Perform differential expression analysis on orthogroups (e.g., with DESeq2) to identify conserved expression patterns associated with specific developmental processes.
Protocol 2: Targeted Sequencing for Deep Interrogation of Gene Families
Objective: To fully characterize all members and variants of a specific gene family (e.g., NLRs in plants or Hox genes in animals) [36].
- Probe or Primer Design: Design biotinylated RNA probes (for hybridization capture) or PCR primers to target the gene family of interest. For long-read sequencing, design primers to generate amplicons that cover the entire genomic locus.
- Library Preparation & Enrichment:
- Hybridization Capture: Prepare a whole-genome sequencing library. Hybridize the library with the custom probes, capture with streptavidin beads, and wash away non-targeted fragments.
- Amplicon Sequencing: Perform long-range PCR using target-specific primers with incorporated barcodes. Purify amplicons and create a PacBio SMRTbell library [36].
- Sequencing: For captured libraries, sequence on an Illumina platform for deep coverage. For amplicons, sequence on a PacBio Sequel IIe system with a 10-20 hour movie time to generate high-fidelity (HiFi) reads.
- Data Analysis:
- Variant Calling: For Illumina data, use GATK for SNP and indel calling. For PacBio HiFi data, use the SMRT Link long amplicon analysis (LAA) workflow to generate highly accurate consensus sequences for each amplicon [36].
- Haplotype Phasing & Structural Variation: Use long-read data to resolve haplotypes and detect structural variations (SVs) within the gene family.
Diagram 1: Generalized NGS workflow for homology detection, showing key stages from sample to biological insight.
Data Analysis: Statistical and Bioinformatic Workflows
The massive datasets generated by NGS require robust statistical and bioinformatic pipelines.
Statistical Considerations for Genome-Wide Data
Analysis of NGS data often involves testing numerous hypotheses simultaneously (e.g., differential expression of tens of thousands of genes, or enrichment of thousands of genetic variants). This necessitates multiple testing corrections (e.g., Bonferroni, Benjamini-Hochberg) to control the false discovery rate (FDR) [41]. Furthermore, data is often analyzed in the form of contingency tables for tasks like gene set enrichment analysis, where Fisher's exact test or chi-squared tests are commonly applied [41].
Key Bioinformatics Tools for Homology Detection
Table 2: Essential Bioinformatics Tools for Homology Detection in NGS Data.
| Tool / Resource | Category | Function in Analysis Workflow |
|---|---|---|
| BLAST/PSI-BLAST [39] | Sequence Alignment | Initial, alignment-based search for homologous sequences. |
| Dense Homolog Retriever (DHR) [40] | Alignment-Free Detection | Rapid, sensitive homology detection using protein language models. |
| OrthoFinder | Orthology Inference | Infers orthogroups and gene trees from whole-genome data. |
| STAR | Read Alignment | Fast, splice-aware alignment of RNA-Seq reads to a reference genome. |
| DESeq2 | Differential Expression | Statistical analysis of differential gene expression from RNA-Seq count data. |
| GATK | Variant Calling | Discovers SNPs and indels in NGS data. |
| SMRT Link (LAA) [36] | Long-Read Analysis | Generates highly accurate consensus sequences from amplicon data. |
| InterPro/Pfam [39] | Domain Annotation | Annotates protein domains and functional sites. |
Diagram 2: A comparative transcriptomics workflow for identifying deep homology across species.
The Scientist's Toolkit: Key Research Reagent Solutions
Table 3: Essential Research Reagents and Kits for NGS-Based Homology Studies.
| Reagent / Kit | Function | Example Application |
|---|---|---|
| Stranded mRNA-Seq Library Prep Kit (e.g., Illumina) | Converts purified mRNA into a sequencing-ready library with strand information. | RNA-Seq for transcriptome assembly and gene expression quantification. |
| Whole Exome Enrichment Kit [38] | Captures and enriches protein-coding regions of the genome using biotinylated probes. | Targeted sequencing for discovering coding variants associated with traits or diseases. |
| SMRTbell Prep Kit [36] | Prepares high-molecular-weight DNA for long-read sequencing on PacBio platforms. | Generating long reads for resolving complex haplotype-phased gene families. |
| Custom Target Enrichment Probes | Biotinylated oligonucleotides designed to capture specific genomic regions of interest. | Deep sequencing of a specific gene regulatory network across multiple species. |
| Barcoded Adapters (e.g., Illumina, PacBio) | Allows for multiplexing of multiple samples in a single sequencing run. | Cost-effective sequencing of numerous individuals or species in a single experiment. |
Next-Generation Sequencing and transcriptomics have provided the technological foundation to elevate the search for homology from a localized, gene-centric endeavor to a systems-level, genome-wide exploration. By leveraging the detailed experimental protocols, sophisticated analytical workflows, and specialized reagent solutions outlined in this guide, researchers can now systematically decode the deep homologous relationships that govern animal design. This approach promises to unravel the conserved genetic logic that has shaped evolutionary diversity, with profound implications for understanding developmental biology, evolutionary history, and the genetic basis of disease.
The prediction of a protein's three-dimensional structure from its amino acid sequence stands as one of the most significant challenges in computational biology. For decades, homology modeling (also known as comparative modeling) has served as a cornerstone technique, operating on the principle that evolutionarily related proteins share similar structures [42] [43]. This foundational concept aligns directly with the broader thesis of deep homology in animal design, which reveals how deeply conserved genetic toolkits and protein architectural plans underlie the vast diversity of animal forms [44] [45]. The remarkable conservation of protein folds across phylogenetically diverse organisms provides the fundamental basis for homology modeling—if a target protein shares sufficient sequence similarity with a template of known structure, a accurate model can be constructed [43] [46].
The paradigm of protein structure prediction has undergone a seismic shift with the advent of deep learning approaches, particularly AlphaFold2 [47] [46]. This transformation does not render homology modeling obsolete but rather redefines its role and implementation within the computational structural biologist's toolkit. Modern deep learning systems have internalized the principles of homology modeling—leveraging evolutionary information from multiple sequence alignments (MSAs) and structural templates—but have done so through neural networks trained on the entire corpus of known protein structures [47] [48]. This review examines the current state of protein structure prediction through the lens of this integration, where the core principles of homology modeling persist, albeit implemented within sophisticated deep learning architectures that have dramatically expanded the scope and accuracy of predictable structures.
Traditional Homology Modeling: Principles and Workflow
Traditional homology modeling operates through a well-established pipeline that transforms a target sequence into a three-dimensional model using experimentally determined structures as templates. The critical dependence on template selection cannot be overstated, with the choice of template(s) often determining the success or failure of the modeling endeavor [44]. The modeling process systematically addresses each component of protein architecture, from the conserved backbone to the variable loop regions and side-chain conformations.
The Homology Modeling Workflow
The standard workflow for homology modeling encompasses several sequential steps, each with specific considerations and potential pitfalls:
Template Identification and Selection: The target sequence is scanned against protein structure databases (primarily the PDB) using search tools like BLAST or PSI-BLAST to identify potential templates [44] [49]. Key selection criteria include sequence identity (typically >30% for reliable models), coverage of the target sequence, and the quality of the template structure (e.g., resolution for X-ray structures) [44].
Target-Template Alignment: The target sequence is aligned with the template structure, establishing the correspondence between amino acids. This step is crucial as alignment errors represent a major source of model inaccuracy [44]. Advanced methods using profile-profile alignments or hidden Markov models can improve accuracy, especially in cases of lower sequence similarity.
Model Building: The core framework of the template structure is used to construct the target protein's backbone. Conserved regions maintain the template's coordinates, while variable regions require specialized approaches [49].
Loop Modeling: Regions with insertions or deletions relative to the template (typically loops) are modeled using database search or de novo techniques, with accuracy decreasing significantly for loops longer than 12-13 residues [44].
Side-Chain Modeling: Non-conserved side chains are added using rotamer libraries that contain statistically favored side-chain conformations observed in high-resolution structures [44] [49]. Tools like SCWRL implement this functionality efficiently [44].
Model Refinement and Validation: The initial model undergoes energy minimization to relieve steric clashes and optimize geometry, followed by quality assessment using validation tools [44]. The MolProbity server provides comprehensive validation including analysis of Ramachandran plots, rotamer outliers, and steric clashes [44].
Table 1: Key Resources for Traditional Homology Modeling
| Resource Type | Examples | Primary Function |
|---|---|---|
| Template Search Tools | BLAST, PSI-BLAST, HHblits | Identify homologous structures for a target sequence [44] [49] |
| Modeling Servers | SWISS-MODEL, Phyre2, I-TASSER | Automated homology modeling pipelines [44] [43] |
| Standalone Software | MODELLER, SCWRL, Rosetta | Customizable modeling and refinement tools [44] [45] |
| Validation Tools | MolProbity, PROCHECK, QMEAN | Assess model quality and identify potential errors [44] [49] |
Figure 1: Traditional homology modeling workflow. The process involves sequential steps from template identification to model validation, with feedback loops for model correction.
Limitations of Traditional Approaches
Despite its utility, traditional homology modeling faces several fundamental limitations. The method is heavily dependent on the availability and quality of suitable templates, making it inapplicable to proteins with novel folds or distant evolutionary relationships [43] [46]. Accuracy decreases sharply when sequence identity falls below 30%, and errors in the initial target-template alignment propagate through subsequent steps, often resulting in structurally compromised models [44]. Furthermore, modeling flexible loop regions and accurately packing side chains remain challenging, particularly for proteins with conformational flexibility or large binding interfaces [50].
The Deep Learning Revolution: AlphaFold2 and Beyond
The development of AlphaFold2 (AF2) by DeepMind represented a paradigm shift in protein structure prediction, achieving accuracy competitive with experimental methods in the 14th Critical Assessment of protein Structure Prediction (CASP14) [47] [46]. Unlike traditional homology modeling, which applies explicit template-based reasoning, AF2 employs an end-to-end deep learning architecture that integrates evolutionary information with physical and geometric constraints of protein structures.
AlphaFold2 Architecture and Innovation
The AF2 network incorporates several groundbreaking innovations that enable its unprecedented performance:
Evoformer Module: A novel neural network block that jointly processes multiple sequence alignments (MSAs) and residue-pair representations, enabling information exchange between evolutionary and structural features [47]. The Evoformer uses attention mechanisms to reason about spatial relationships and evolutionary constraints simultaneously.
Structure Module: This component generates atomic coordinates through iterative refinement, starting from initial states and progressively developing precise protein structures with accurate atomic details [47]. It employs a rotation-equivariant architecture that respects the physical symmetries of molecular structures.
Recycling and Iterative Refinement: The system repeatedly processes its own outputs through the same network modules, allowing progressive refinement of structural hypotheses [47]. This recursive approach mimics the iterative nature of traditional modeling but occurs within a single forward pass of the network.
Self-Distillation and Confidence Estimation: AF2 incorporates a self-estimate of accuracy (pLDDT) for each residue, enabling users to identify reliable regions of the prediction [47]. The model was also trained on its own high-quality predictions, expanding its knowledge beyond experimentally determined structures.
Figure 2: AlphaFold2's core architecture. The system processes sequence and evolutionary information through the Evoformer and Structure modules, with iterative refinement via recycling.
Beyond AlphaFold2: Recent Advancements
Since the release of AlphaFold2, the field has continued to evolve with new systems addressing specific limitations. AlphaFold3 has extended capabilities to predict complexes containing proteins, nucleic acids, and ligands [51]. RoseTTAFold implements a similar three-track architecture but with reduced computational requirements [46]. Most notably, DeepSCFold represents a significant advance for protein complex prediction, using sequence-derived structural complementarity to achieve improvements of 11.6% and 10.3% in TM-score compared to AlphaFold-Multimer and AlphaFold3 respectively on CASP15 targets [51].
For antibody-antigen complexes—historically challenging due to limited co-evolutionary signals—DeepSCFold enhances the prediction success rate for binding interfaces by 24.7% and 12.4% over AlphaFold-Multimer and AlphaFold3, respectively [51]. This demonstrates how specialized architectures can address specific biological questions beyond general protein folding.
Integration and Comparison: Quantitative Assessment
The relationship between traditional homology modeling and modern deep learning approaches is not one of replacement but rather integration and enhancement. Deep learning systems have internalized the principles of homology modeling while overcoming many of its limitations through learned representations of structural space.
Table 2: Performance Comparison of Structure Prediction Methods
| Method | Approach | Typical TM-score* / GDT_TS | Key Applications | Limitations |
|---|---|---|---|---|
| Traditional Homology Modeling | Template-based structure construction | Varies with template quality (GDT_TS: >80% for >50% sequence identity) [46] | Proteins with close homologs; quick preliminary models [44] | Template dependence; poor for novel folds [43] |
| AlphaFold2 | Deep learning with MSAs and templates | 0.8-0.9 TM-score (high accuracy range) [47] | High-accuracy monomer predictions; structural genomics [46] | Computational intensity; limited conformational diversity [48] |
| AlphaFold-Multimer | AF2 adapted for complexes | Lower than AF2 for monomers [51] | Protein-protein complexes [51] | Reduced accuracy compared to monomer AF2 [51] |
| DeepSCFold | Sequence-derived structure complementarity | 11.6% improvement over AF-Multimer in TM-score [51] | Challenging complexes (antibody-antigen) [51] | Specialized for complexes; newer method with less testing |
TM-score: >0.5 indicates correct fold, >0.8 high accuracy
Practical Performance Considerations
While deep learning methods have demonstrated superior accuracy overall, traditional homology modeling retains value in specific scenarios. For proteins with very close templates (>50% sequence identity), homology modeling can produce models comparable in accuracy to AF2 but with significantly reduced computational requirements [46]. Additionally, the explicit nature of traditional modeling allows for greater manual intervention and expert curation, which remains valuable for addressing problematic regions or incorporating experimental constraints [44] [49].
The performance advantage of deep learning methods becomes most pronounced in the "twilight zone" of sequence similarity (<30% identity), where traditional methods struggle but AF2 frequently produces accurate models [47] [46]. This capability has dramatically expanded the scope of protein families for which reliable models can be generated, directly advancing studies of deep homology by revealing structural conservation across distantly related taxa.
The Scientist's Toolkit: Research Reagent Solutions
Table 3: Essential Resources for Modern Protein Structure Prediction
| Resource | Type | Key Features | Application Context |
|---|---|---|---|
| AlphaFold2/ColabFold | Deep Learning Server | State-of-the-art accuracy; user-friendly interface [50] [46] | High-accuracy monomer predictions; rapid prototyping |
| SWISS-MODEL | Homology Modeling Server | Automated pipeline; reliable for template-based modeling [44] [49] | Quick models when close templates available |
| RoseTTAFold | Deep Learning Server | Three-track architecture; lower computational demand [46] | Balanced accuracy and efficiency |
| DeepSCFold | Specialized Deep Learning | Structure complementarity for complexes [51] | Challenging protein-protein interactions |
| MODELLER | Standalone Software | Customizable modeling; scriptable pipeline [44] [45] | Incorporating experimental constraints; specialized applications |
| PDB | Structure Repository | Source of templates; experimental validation [44] [45] | Template selection; model validation |
| UniRef/UniProt | Sequence Databases | Source for MSA construction [51] [46] | Evolutionary information for deep learning methods |
Experimental Protocols and Methodologies
Protocol: Traditional Homology Modeling with SWISS-MODEL
The SWISS-MODEL pipeline provides a robust implementation of traditional homology modeling principles, accessible through both web interface and programmatic access [49]:
Input Preparation: Provide the target amino acid sequence in FASTA format or as a UniProtKB accession code. Alternatively, upload a custom target-template alignment for alignment mode.
Template Selection: The server performs automated template identification using BLAST and HHblits against the SWISS-MODEL Template Library (SMTL). Templates are ranked by sequence identity, coverage, and quality measures.
Model Building: The ProMod3 modeling engine constructs the model using coordinates from conserved regions, database search for loops, and backbone-dependent rotamer libraries for side chains [49].
Quality Assessment: Models are evaluated using QMEAN scoring, which combines statistical potentials of mean force to assess global and local model quality [49].
Ligand and Oligomer Modeling: Biologically relevant ligands are transferred from templates if coordinating residues are conserved. Quaternary structure is modeled using a support vector machine-based algorithm that estimates quaternary structure quality (QSQE) [49].
Protocol: Deep Learning-Assisted Complex Prediction with DeepSCFold
DeepSCFold represents the integration of homology modeling principles with deep learning for challenging prediction tasks [51]:
Input Processing: Provide sequences of interacting protein chains. Generate monomeric multiple sequence alignments (MSAs) from standard databases (UniRef30, UniProt, etc.).
Structural Similarity Prediction: A deep learning model predicts protein-protein structural similarity (pSS-score) between query sequences and their homologs, enhancing MSA ranking beyond sequence similarity.
Interaction Probability Estimation: A second deep learning model predicts interaction probability (pIA-score) between sequence homologs from different subunit MSAs.
Paired MSA Construction: Monomeric homologs are systematically concatenated using interaction probabilities to construct biologically relevant paired MSAs.
Complex Structure Prediction: The series of paired MSAs are used by AlphaFold-Multimer to generate complex structures, with model selection via quality assessment methods.
Template-Based Refinement: The top model serves as an input template for a final iteration of AlphaFold-Multimer to produce the output structure [51].
The field of protein structure prediction has undergone a remarkable transformation, with deep learning systems internalizing and extending the principles of homology modeling. The integration of these approaches has created a powerful paradigm where evolutionary information from MSAs and structural templates is processed through learned representations of structural space. This synergy has been particularly valuable for investigating deep homology in animal design, as it enables accurate structural modeling of conserved protein domains across diverse taxa.
Future advancements will likely focus on several key areas: improving predictions for complex molecular assemblies including membrane proteins and nucleic acid complexes; enhancing accuracy for conformationally flexible systems; and developing efficient methods that reduce computational requirements while maintaining accuracy [48]. As these methods continue to evolve, they will further illuminate the deep structural homologies underlying biological diversity, providing unprecedented insights into the molecular basis of animal design and function.
The identification of remote homologs represents a fundamental challenge in computational biology, bridging the gap between sequence information and deep functional and evolutionary relationships. This whitepaper provides an in-depth technical examination of three powerful methodologies—Deep Homology Recognition (DHR), PSI-BLAST, and HMMER—for detecting distant evolutionary relationships in protein sequences. Framed within the context of deep homology in animal design research, this guide details the operational principles, comparative performance metrics, and practical implementation protocols for each tool. By synthesizing current research and providing structured comparisons and visual workflows, this document serves as a comprehensive resource for researchers, scientists, and drug development professionals seeking to elucidate ancient evolutionary patterns that inform modern animal design principles.
Deep homology refers to the conservation of genetic regulatory circuits and protein architectures across vastly divergent animal lineages, often underlying similar morphological structures despite extensive sequence divergence. The detection of remote homology—evolutionary relationships that have become obscured at the sequence level—is therefore critical to understanding the fundamental building blocks of animal design. Traditional sequence comparison methods like BLAST excel at identifying close homologs but lack sensitivity for detecting distant evolutionary connections that persist in structural and functional domains.
Profile-based methods significantly enhance detection sensitivity by exploiting conservation patterns within multiple sequence alignments. Among these, PSI-BLAST (Position-Specific Iterative BLAST) and HMMER (utilizing profile Hidden Markov Models) represent established computational approaches that have demonstrated superior performance over pairwise methods for detecting remote homology [52]. More recently, Deep Homology Recognition (DHR) approaches leveraging deep learning architectures have emerged to address even more challenging relationships. These tools collectively enable researchers to traverse deeper evolutionary time, revealing homologous relationships that inform our understanding of how ancient genetic elements have been co-opted and repurposed throughout animal evolution.
Core Methodologies and Operational Principles
PSI-BLAST (Position-Specific Iterative BLAST)
PSI-BLAST operates through an iterative search process that progressively refines a position-specific scoring matrix (PSSM) to capture increasingly subtle conservation patterns. The fundamental workflow begins with a standard BLASTp search of a query sequence against a protein database. Significant hits from this initial search are incorporated into a multiple sequence alignment, from which a PSSM is derived. This PSSM, which encapsulates position-specific conservation information, then replaces the original query for a subsequent database search. This process iterates until convergence (no new significant hits are detected) or a specified iteration limit is reached [53].
The power of PSI-BLAST lies in its ability to combine conservation information from multiple related sequences into a single scoring matrix, enabling detection of sequence similarities that are undetectable through pairwise methods alone. This approach has proven particularly valuable for identifying relationships between proteins with conserved three-dimensional structures despite considerable sequence erosion [53]. For example, PSI-BLAST can detect the distant relationship between eukaryotic proliferating cell nuclear antigen (PCNA) and the Escherichia coli DNA polymerase III β-subunit—structurally similar sliding DNA clamps that perform analogous functions in DNA replication but are undetectable via conventional BLAST [53].
HMMER (Profile Hidden Markov Models)
The HMMER package implements profile hidden Markov models for sensitive biological sequence analysis. The profile HMM procedure involves three distinct stages: (1) construction of a multiple sequence alignment of known members of a protein family, (2) building a profile HMM from this alignment incorporating prior knowledge of protein generalities, and (3) scoring query sequences against the model to determine family membership probability [52]. The quality of the initial multiple sequence alignment and the diversity of sequences it contains are crucial determinants of the final model's discriminatory power.
HMMER employs statistical methods to calibrate models against random sequences, fitting extreme value distributions to raw scores to calculate accurate E-values for database searches [52]. Compared to PSI-BLAST, profile HMMs generally offer greater sensitivity for detecting remote homologs, particularly when starting from carefully curated multiple sequence alignments. The SAM package provides an alternative HMM implementation with its T99 script for automated alignment generation, which has been shown to produce high-quality alignments that contribute to improved model performance [52].
Deep Homology Recognition (DHR)
Deep Homology Recognition represents an emerging approach leveraging deep learning architectures to identify distant evolutionary relationships that evade traditional methods. While specific implementations vary, DHR typically employs deep neural networks with attention mechanisms or convolutional layers to automatically learn discriminative features from sequence or alignment data without relying on handcrafted features. These systems can identify subtle patterns and contextual relationships across multiple sequence scales that correlate with deep homologous relationships.
The DeepHL platform exemplifies this approach, using a deep neural network based on an attention mechanism to automatically detect characteristic segments in trajectories or sequences that differentiate between groups [54]. Although initially developed for comparative analysis of animal movement data, similar architectural principles apply to protein sequence analysis. These networks transform input sequences into time series of features (e.g., position-specific conservation metrics), which are processed through multi-scale layer-wise attention models to identify regions of maximal discriminative power [54].
Comparative Performance Analysis
Sensitivity and Specificity
Studies comparing profile HMM methods have demonstrated that SAM consistently produces better models than HMMER from identical alignments when using default parameters, though relative performance of model-scoring components varies [52]. The critical determinant of overall profile HMM performance is the quality of multiple sequence alignments used for model construction. The SAM T99 iterative database search procedure, which automatically generates high-quality alignments, performs better than the most recent version of PSI-BLAST at the time of the study [52].
Performance evaluations using structurally informed benchmarks like SCOP have confirmed the superiority of profile-based methods over pairwise approaches for detecting remote homology at family and superfamily levels [52]. Hybrid approaches that combine HMMs with PSI-BLAST can further enhance detection sensitivity, as demonstrated by studies where PSI-BLAST searches initialized with profiles derived from HMMs revealed distant structural relationships with substantially greater sensitivity than standard PSI-BLAST in certain instances [55].
Table 1: Comparative Performance Metrics of Remote Homology Detection Tools
| Tool | Methodology | Primary Strength | Optimal Use Case | Detection Range |
|---|---|---|---|---|
| PSI-BLAST | Position-Specific Iterative Matrix | Speed, efficiency | Finding distant homologs with some sequence conservation | Moderate distance |
| HMMER | Profile Hidden Markov Models | Sensitivity with good alignments | Domain detection, pre-defined families | Distant relationships |
| SAM | Profile HMMs with T99 | Automated alignment generation | Full automation from single sequence | Distant relationships |
| DHR | Deep Learning | Feature learning without pre-definition | Novel relationship discovery | Very distant relationships |
Computational Efficiency
Benchmarking studies reveal significant differences in computational requirements between methods. HMMER is typically between one and three times faster than SAM when searching databases larger than 2000 sequences, though SAM demonstrates advantages for smaller databases [52]. PSI-BLAST profile scoring is notably efficient—more than 30 times faster than scoring SAM models—making it particularly suitable for large-scale analyses [52].
Both HMMER and SAM implement effective low-complexity and repeat sequence masking using their null models, with comparable accuracy in E-value estimation [52]. The computational intensity of deep learning-based DHR approaches varies considerably with model architecture, with convolutional networks generally offering faster processing than recurrent or attention-based models, though all typically require greater resources than traditional methods.
Table 2: Computational Requirements and Practical Considerations
| Tool | Speed | Memory Usage | Ease of Use | Automation Potential |
|---|---|---|---|---|
| PSI-BLAST | Very Fast | Low | Moderate (iteration management) | High with careful thresholding |
| HMMER | Fast (large DB) / Moderate | Moderate | Moderate (alignment dependent) | High with good initial alignment |
| SAM | Slow (large DB) / Fast | High | High (with T99 automation) | Very high |
| DHR | Variable (often slow) | Very High | Low (expert tuning needed) | Moderate once trained |
Experimental Protocols and Implementation
PSI-BLAST Practical Protocol
Objective: Identify distant homologs of a query protein using iterative profile refinement.
Materials:
- Query protein sequence (FASTA format)
- NCBI NR database or specialized protein database
- Computing resources with PSI-BLAST installed or web access
Methodology:
- Initialization: Submit query sequence to PSI-BLAST via web interface (http://www.ncbi.nlm.nih.gov/BLAST/) or command line.
- Parameter Configuration:
- Set database to "nr" for comprehensive search
- Set E-value threshold to 0.005 for profile inclusion (beginners) or 0.01 (experienced users)
- Adjust maximum target sequences to 1000 to ensure comprehensive retrieval
- Iteration:
- Execute first iteration (standard BLASTp)
- Review hits meeting inclusion threshold
- Generate multiple alignment and PSSM automatically
- Execute subsequent iterations using "Run next iteration"
- Continue until convergence (no new hits) or 5-10 iterations
- Validation:
- Manually inspect alignments of putative homologs
- Verify conservation patterns match structural/functional expectations
- Check for transitive identification errors
Technical Notes: For sequences with compositionally biased segments, adjust inclusion thresholds more stringently to minimize false positives. The E. coli DNA polymerase III β-subunit example required five iterations to detect relationship with PCNA using human PCNA as query [53].
HMMER Model Construction and Searching
Objective: Build a profile HMM from a multiple sequence alignment and search for remote homologs.
Materials:
- Multiple sequence alignment of protein family (Stockholm, FASTA, or aligned format)
- Sequence database for searching
- HMMER software suite (http://hmmer.wustl.edu)
Methodology:
- Alignment Preparation:
- Curate multiple sequence alignment with representative family members
- Ensure proper alignment of conserved domains and motifs
- Model Construction:
- Execute:
hmmbuild model.hmm alignment.sto - Model calibration:
hmmpress model.hmm
- Execute:
- Database Searching:
- Execute:
hmmscan model.hmm database.fasta - Review E-values and domain architecture of hits
- Execute:
- Iterative Refinement:
- Add confident hits to alignment
- Rebuild model and repeat search
- Validate with known negatives
Technical Notes: HMMER calibration fits extreme value distributions to scores against random sequences to calculate accurate E-values. Model performance is highly dependent on alignment quality and sequence diversity [52]. The globin and cupredoxin family analyses demonstrate the critical importance of alignment quality on eventual model sensitivity [52].
Deep Homology Recognition Implementation
Objective: Apply deep learning to identify distant homologous relationships without predefined features.
Materials:
- Curated training set of homologous and non-homologous sequences
- Computational resources with deep learning framework (TensorFlow, PyTorch)
- DHR implementation or architecture specifications
Methodology:
- Data Preparation:
- Convert sequences to feature representations (e.g., PSSM, one-hot encoding)
- Partition into training, validation, and test sets
- Ensure balanced class representation
- Model Configuration:
- Implement multi-scale architecture with attention mechanisms
- Configure convolutional layers for local feature extraction
- Include LSTM layers for long-range dependency modeling
- Training:
- Train with backpropagation using discriminative loss function
- Monitor validation performance for early stopping
- Apply regularization to prevent overfitting
- Interpretation:
- Extract attention weights to identify discriminative sequence regions
- Correlate with known structural and functional features
Technical Notes: The DeepHL framework demonstrates how attention mechanisms can highlight characteristic segments in trajectories [54]. Similar principles apply to protein sequences, where attention weights identify residues and regions most informative for discriminating between homologous groups.
Visualization and Analytical Workflows
Remote Homology Detection Workflow
The following diagram illustrates the integrated workflow for remote homology detection using the three complementary approaches discussed, highlighting decision points and optimal tool selection based on available data and research objectives:
PSI-BLAST Iterative Search Mechanism
The iterative search mechanism of PSI-BLAST represents a powerful approach for progressively detecting more distant homologs through profile refinement:
Research Reagent Solutions
Table 3: Essential Computational Tools and Resources for Remote Homology Detection
| Tool/Resource | Type | Primary Function | Access | Application Context |
|---|---|---|---|---|
| NCBI NR Database | Database | Comprehensive non-redundant protein sequences | https://www.ncbi.nlm.nih.gov/ | Primary search database for PSI-BLAST |
| Pfam Database | Database | Curated protein families and domains | http://pfam.xfam.org/ | HMMER model source and validation |
| HMMER Software Suite | Software | Profile HMM construction and searching | http://hmmer.wustl.edu/ | Remote homology detection from alignments |
| SAM-T99 | Algorithm | Automated multiple sequence alignment generation | http://compbio.soe.ucsc.edu/sam.html | Alignment construction for HMMER |
| DeepHL Framework | Software | Deep learning-based comparative analysis | Reference implementation [54] | Pattern discovery in sequence features |
| SCOP Database | Database | Structural classification of proteins | http://scop.mrc-lmb.cam.ac.uk/ | Validation and benchmark testing |
Applications in Animal Design Research
The detection of remote homology has profound implications for understanding animal design principles. Deep homology concepts suggest that ancient genetic circuits and protein architectures have been conserved and repurposed throughout animal evolution, generating diverse morphological structures from common genetic foundations. Remote homology detection tools enable researchers to trace these deep evolutionary relationships, revealing how:
Conserved developmental regulators such as Hox genes and signaling pathway components have maintained core functions despite sequence divergence across animal phyla.
Protein domain architectures associated with specific cellular functions (e.g., cell adhesion, neural signaling) show unexpected conservation between distantly related organisms.
Gene regulatory networks controlling fundamental developmental processes often share deeply homologous components across bilaterians.
PSI-BLAST has demonstrated particular utility in identifying relationships between structurally similar proteins with divergent sequences, such as the sliding DNA clamps involved in DNA replication across eukaryotes, prokaryotes, and viruses [53]. HMMER's sensitivity makes it ideal for identifying conserved domains in novel animal genes that may represent previously unrecognized members of established gene families. DHR approaches offer potential for discovering entirely novel types of homologous relationships that do not conform to established domain architectures.
The revolutionary detection tools DHR, PSI-BLAST, and HMMER provide complementary approaches for identifying remote homologs critical to understanding deep homology in animal design. While each method offers distinct advantages—PSI-BLAST for its efficiency and iterative refinement, HMMER for its sensitivity with quality alignments, and DHR for its potential to discover novel relationships—their combined application offers the most powerful strategy for elucidating evolutionary connections across deep time.
Future developments will likely focus on integrating these approaches into unified pipelines, improving the automation of high-quality multiple sequence alignment construction, and enhancing the interpretability of deep learning-based methods. As genomic data continue to expand across the tree of life, these tools will become increasingly essential for deciphering the deep homologies that underlie animal design principles, ultimately informing both evolutionary biology and biomedical research through the identification of conserved functional modules with therapeutic potential.
The integration of deep homology into the drug discovery pipeline represents a paradigm shift in target identification and validation. By leveraging evolutionarily conserved genetic circuits and developmental modules across species, researchers can now identify druggable targets with enhanced precision and biological relevance. This technical guide elucidates how the principles of deep homology—the remarkable conservation of genetic toolkits governing morphological development across distantly related organisms—are being operationalized through advanced computational frameworks to accelerate pharmaceutical development. We present comprehensive methodologies, performance benchmarks, and practical protocols for implementing deep homology-based approaches, demonstrating their transformative potential in addressing the critical challenges of cost, timeline, and efficacy in modern drug development.
Deep homology transcends traditional sequence-based homology by recognizing that despite vast evolutionary divergence and absence of phylogenetic continuity, distantly related species often utilize remarkably conserved genetic circuitry during development [8]. This concept originated from evolutionary developmental biology (evo-devo) findings that seemingly novel anatomical features in different phyla frequently develop from homologous developmental modules. For instance, despite 600 million years of evolutionary separation, the same genetic regulatory networks govern appendage development in both insects and vertebrates, demonstrating that deep homology operates at the level of core genetic circuits rather than structural outcomes [8].
The mechanistic foundation of deep homology lies in what has been termed "character identity networks" (ChINs)—core gene regulatory networks (GRNs) that define the developmental identity of specific morphological structures [8]. Unlike sequence homology, which depends on historical continuity and percentage identity, deep homology manifests through the conserved deployment of these regulatory kernels across vastly different organisms. This conceptual framework provides a powerful lens for identifying biologically critical targets in drug discovery, as these deeply conserved networks often represent fundamental biological processes whose disruption leads to disease.
Computational Frameworks for Deep Homology Detection
Advanced Algorithmic Approaches
The detection and exploitation of deep homology require computational methods capable of identifying structural and functional similarities beyond sequence-level comparisons. Several innovative frameworks have demonstrated significant efficacy in this domain:
TM-Vec and DeepBLAST represent breakthrough methodologies for protein remote homology detection. TM-Vec employs a twin neural network architecture trained to approximate TM-scores (metrics of structural similarity) directly from sequence pairs without requiring intermediate structure computation [5]. This enables scalable structure-aware searching across massive protein databases. DeepBLAST performs structural alignments using differentiable dynamic programming and protein language models, predicting structural alignments analogous to those generated by TM-align but using only sequence information [5]. When validated on CATH protein domains clustered at 40% sequence similarity, TM-Vec maintained high prediction accuracy (r = 0.936, P < 1 × 10⁻⁵) even for sequences with less than 0.1% identity, far surpassing traditional sequence alignment capabilities [5].
optSAE + HSAPSO integrates stacked autoencoders for robust feature extraction with hierarchically self-adaptive particle swarm optimization for adaptive parameter tuning [56]. This framework specifically addresses limitations of traditional machine learning approaches in handling complex pharmaceutical datasets, achieving 95.52% accuracy in drug classification and target identification on DrugBank and Swiss-Prot datasets while significantly reducing computational complexity to 0.010 seconds per sample [56].
Top-DTI combines topological deep learning with large language models to predict drug-target interactions by integrating persistent homology features from protein contact maps with semantically rich embeddings from protein sequences and drug SMILES strings [57]. This approach has demonstrated superior performance in challenging cold-split scenarios where test sets contain drugs or targets absent from training data, closely simulating real-world discovery environments [57].
Table 1: Performance Benchmarks of Deep Homology Detection Frameworks
| Framework | Core Methodology | Accuracy Metrics | Specialized Capabilities |
|---|---|---|---|
| TM-Vec | Twin neural networks predicting TM-scores from sequences | r=0.936 on CATH domains; 99.9% true positive rate for fold prediction | Scalable structural similarity search; functions below 0.1% sequence identity |
| optSAE+HSAPSO | Stacked autoencoder with adaptive particle swarm optimization | 95.52% accuracy; 0.010s/sample computational speed | High stability (±0.003); reduced overfitting in high-dimensional data |
| Top-DTI | Topological data analysis + large language models | Superior AUROC, AUPRC in cold-split scenarios | Robust performance on novel targets absent from training data |
| AiCDR | Generative adversarial network with dual external discriminators | Experimentally validated nanobody neutralization | Natural-like CDR3 sequence generation for therapeutic antibody design |
Experimental Protocol: Implementing TM-Vec for Remote Homology Detection
Objective: Identify structurally similar proteins with potential functional homology despite low sequence similarity.
Materials and Computational Requirements:
- Hardware: GPU-enabled computational node (minimum 16GB VRAM)
- Software: Python 3.8+, PyTorch 1.10+, TM-Vec implementation
- Databases: SWISS-MODEL, CATH, or custom protein sequence databases
Methodology:
- Database Preprocessing: Convert protein sequences to FASTA format and remove redundant entries at 90% identity threshold.
- Model Initialization: Load pre-trained TM-Vec weights or train de novo on structural database (277,000+ SWISS-MODEL chains).
- Embedding Generation: Process sequences through the twin neural network to generate structure-aware vector embeddings.
- Similarity Search: Query target protein against embedded database using cosine distance metric (approximates TM-score).
- Validation: For top candidates (cosine distance >0.7, approximating TM-score >0.5), perform structural confirmation with DeepBLAST alignment.
- Functional Annotation Transfer: Infer potential functional homology for targets sharing structural similarity (TM-score >0.5) despite low sequence identity.
Interpretation Guidelines:
- TM-score >0.5: Same fold, high confidence in functional homology
- TM-score 0.3-0.5: Similar folds, moderate confidence
- TM-score <0.3: Different folds, low confidence
- Sequence identity <25% with TM-score >0.5: Indicator of deep homology
Deep Homology in Target Identification and Validation
Paradigm Shifts in Druggable Target Identification
The integration of deep homology principles has fundamentally transformed target identification from sequence-based to systems-based approaches. Traditional methods like BLAST and PSI-BLAST effectively identify close homologs but fail at evolutionary distances beyond 25% sequence identity—precisely where deep homology approaches excel [5]. By focusing on structural conservation and functional GRN preservation, researchers can now identify targets across several critical domains:
Disease Modeling and Target Prioritization: Deep homology facilitates the identification of evolutionarily conserved pathways central to disease pathogenesis. For rare diseases, where patient data is scarce, deep homology enables triangulation between model organisms and human biology. Computational studies using Orphanet data suggest that borderline-common disorders involve more complex genetic architectures than ultra-rare diseases, underscoring the value of integrative genome-phenome modeling informed by deep homology principles [58].
Therapeutic Antibody Design: The AiCDR framework demonstrates how deep generative modeling can leverage conserved structural principles to design nanobodies targeting specific epitopes [59]. By grafting generated CDR3 sequences onto humanized scaffolds, researchers created a library of 5,200 high-confidence nanobody models, with epitope profiling showing strong overlap with known functional regions across six protein targets [59]. Experimental validation confirmed that two computationally designed nanobodies targeting SARS-CoV-2 Omicron RBD exhibited detectable neutralization activity, demonstrating the practical utility of this approach [59].
Network Pharmacology and Polypharmacology: Deep homology enables the mapping of conserved regulatory kernels across biological systems, facilitating the identification of targets with optimal therapeutic profiles. Rather than single-target approaches, AI-based discovery tools are increasingly shifting toward systems-level modeling of drug-gene-phenotype interactions, enhancing their relevance for diseases with poorly characterized pathophysiology [58].
Experimental Protocol: optSAE+HSAPSO for Target Classification
Objective: Classify and validate druggable targets using stacked autoencoders with adaptive optimization.
Materials:
- Dataset: Curated pharmaceutical data (DrugBank, Swiss-Prot)
- Software: Python implementation of optSAE+HSAPSO
- Computational resources: Multi-core CPU system (minimum 32GB RAM)
Methodology:
- Data Preprocessing:
- Extract protein features (sequence, structural, physicochemical properties)
- Normalize features using z-score transformation
- Split dataset: 70% training, 15% validation, 15% testing
Model Configuration:
- Initialize stacked autoencoder with 5 hidden layers (dimensions: 1024, 512, 256, 512, 1024)
- Configure HSAPSO with 50 particles, cognitive parameter c1=1.5, social parameter c2=1.8
- Set adaptive inertia weight (w=0.9-0.4) based on iteration progress
Training Protocol:
- Pre-train autoencoder layers individually (unsupervised)
- Fine-tune entire network with backpropagation
- Simultaneously optimize hyperparameters using HSAPSO
- Termination condition: Validation loss plateaus for 50 consecutive iterations
Validation and Interpretation:
- Evaluate on held-out test set using accuracy, precision, recall, F1-score
- Perform feature importance analysis to identify critical target characteristics
- Compare against traditional methods (SVM, XGBoost, Random Forest)
Technical Notes:
- Implementation detail: HSAPSO dynamically adjusts exploration-exploitation balance during training
- Critical parameter: Learning rate adaptation based on particle fitness variance
- Advantage: Significantly reduced computational overhead compared to grid search
Visualization Frameworks for Deep Homology Analysis
Deep Homology Detection Workflow
Conserved Regulatory Kernels in Drug Targeting
Research Reagent Solutions for Deep Homology Studies
Table 2: Essential Research Reagents and Computational Tools
| Reagent/Tool | Function | Application in Deep Homology |
|---|---|---|
| TM-Vec | Protein structural similarity prediction from sequence | Remote homology detection beyond sequence identity thresholds |
| DeepBLAST | Structural alignment from sequence information | Validation of detected homologies and functional inference |
| optSAE+HSAPSO | Feature extraction and adaptive parameter optimization | High-accuracy classification of druggable targets |
| AiCDR | Generative nanobody design with epitope profiling | Therapeutic antibody development against conserved epitopes |
| CATH Database | Curated protein domain classification | Benchmarking and validation of homology detection methods |
| SWISS-MODEL | Protein structure homology modeling | Template database for structural feature extraction |
| Top-DTI | Topological deep learning for drug-target prediction | Integration of structural and sequential information for interaction prediction |
The integration of deep homology principles into the drug discovery pipeline represents a fundamental advancement in target identification and validation. By transcending the limitations of sequence-based homology through structural conservation and regulatory network preservation, these approaches enable researchers to identify biologically relevant targets with higher precision and confidence. The computational frameworks outlined in this guide—TM-Vec, optSAE+HSAPSO, Top-DTI, and AiCDR—demonstrate robust performance across multiple metrics, significantly outperforming traditional methods in scenarios with limited sequence similarity.
As the field evolves, several emerging trends promise to further enhance the utility of deep homology in pharmaceutical development. The integration of large language models with topological data analysis, as exemplified by Top-DTI, represents a particularly promising direction [57]. Similarly, the successful experimental validation of computationally designed nanobodies targeting SARS-CoV-2 demonstrates the translational potential of these approaches [59]. Future advancements will likely focus on multi-scale modeling integrating deep homology principles with quantitative systems pharmacology and organ-on-chip technologies, potentially enabling comprehensive in silico simulation of drug effects across biological scales from genetic regulatory networks to organism-level pathophysiology.
The era of deep homology-driven drug discovery is positioned to substantially address the critical challenges of cost, timeline, and efficacy that have long plagued pharmaceutical development. By leveraging the deep evolutionary conservation of biological systems, researchers can now identify targets with greater biological relevance, design therapeutics with optimized interaction profiles, and accelerate the translation of basic biological insights into clinically effective therapies.
Structure-Based Drug Design (SBDD) represents a cornerstone of modern pharmaceutical development, enabling researchers to rationally design therapeutic compounds based on the three-dimensional structure of biological targets. Within this paradigm, homology modeling serves as a critical computational technique when experimental protein structures are unavailable, providing atomic-scale models that guide lead optimization and Structure-Activity Relationship (SAR) analysis. Also known as comparative modeling, homology modeling predicts the three-dimensional structure of a target protein based on its amino acid sequence alignment with evolutionarily related proteins whose structures have been experimentally determined [60]. This approach is particularly valuable for addressing historically "undruggable" targets and for projects where obtaining high-resolution crystal structures proves challenging or time-consuming.
The integration of homology modeling into the drug discovery workflow aligns with the broader concept of deep homology—the evolutionary conservation of protein structural folds and functional mechanisms across diverse species. This conservation enables researchers to leverage the growing repository of structural biology data to build reliable models for previously uncharacterized targets. As the field advances, homology modeling has evolved from a technique of last resort to a robust, predictive tool that significantly accelerates the hit-to-lead optimization process, especially when complemented by emerging artificial intelligence approaches [61]. This technical guide examines the methodologies, applications, and best practices for utilizing homology models in lead optimization and SAR studies, providing drug development professionals with a comprehensive framework for implementing these approaches in their research programs.
Theoretical Foundations and Methodological Framework
Fundamental Principles of Homology Modeling
Homology modeling operates on the fundamental principle that protein structure is more conserved than sequence during evolution. Consequently, even proteins with relatively low sequence similarity often share similar three-dimensional folds if they are evolutionarily related. The accuracy of a homology model depends primarily on the sequence identity between the target and template proteins; generally, sequence identity above 30% typically yields models with reliable backbone structures, while identities above 50% produce models accurate enough for most drug design applications [60]. The modeling process involves a series of sequential steps that progressively refine the target structure, from initial alignment to final model validation.
The methodological framework for homology modeling incorporates both traditional sequence-based algorithms and modern deep learning approaches. Traditional methods like MODELLER implement satisfaction of spatial restraints derived from the template structure, while contemporary tools such as AlphaFold leverage deep learning networks trained on known structures from the Protein Data Bank [60] [62]. These complementary approaches can be integrated to maximize model accuracy, particularly for challenging targets with limited template availability or unusual structural features.
Comparative Analysis of Protein Structure Prediction Methods
Table 1: Comparison of Computational Modeling Approaches for Protein Structure Prediction
| Method | Theoretical Basis | Best Application Context | Advantages | Limitations |
|---|---|---|---|---|
| Homology Modeling | Comparative modeling using evolutionary related templates | High sequence identity (>30%) to known structures [60] | Fast computation; reliable for conserved regions [60] | Template dependence; accuracy decreases with lower sequence identity [60] |
| Threading | Fold recognition using structural templates | Moderate sequence identity; hydrophobic peptides [60] | Identifies distant homologs; useful for novel folds | Limited by template library coverage; alignment challenges |
| AlphaFold | Deep learning on known structures | Single-chain proteins; no template available [62] | High accuracy even without templates [60] | Limited performance on short peptides; requires substantial computational resources [60] |
| PEP-FOLD | De novo assembly from fragments | Short peptides (<50 aa); hydrophilic peptides [60] | No template needed; optimized for short sequences | Limited accuracy for longer sequences; conformational sampling challenges |
Integrated Workflow: From Sequence to Optimized Lead
Homology Modeling Protocol
The construction of a reliable homology model follows a systematic workflow that transforms sequence information into a validated three-dimensional structure. The following diagram illustrates this comprehensive process:
Step 1: Template Identification and Selection The process initiates with a comprehensive search for suitable template structures using sequence similarity search tools like BLAST or PSI-BLAST against the Protein Data Bank (PDB). Selection criteria prioritize templates with high sequence identity, superior resolution (preferably <2.0 Å), and relevant biological context (e.g., similar ligands or bound states) [63]. Multiple templates may be selected for different domains or regions of the target protein to maximize model quality.
Step 2: Sequence Alignment and Backbone Generation Precise sequence alignment between the target and template constitutes the most critical determinant of final model accuracy. Advanced alignment algorithms incorporating secondary structure prediction and evolutionary information often outperform simple pairwise methods. The backbone coordinates from conserved regions of the template are then transferred to the target sequence, establishing the fundamental structural framework [60].
Step 3: Loop Modeling and Side Chain Optimization Non-conserved loop regions, particularly those with insertions or deletions relative to the template, require specialized modeling approaches such as ab initio fragment assembly or database mining. Side chains are initially placed using rotamer libraries that prioritize statistically favored conformations, followed by energy minimization to resolve steric clashes and optimize side chain packing [60].
Step 4: Model Refinement and Validation Molecular dynamics simulations and energy minimization techniques refine the initial model to improve stereochemistry and relieve structural strain. Validation employs multiple metrics including Ramachandran plot analysis (assessing backbone dihedral angles), residue interaction geometry, and comparison with expected structural features. Models passing validation thresholds proceed to drug discovery applications, while failures necessitate template re-evaluation or alternative modeling approaches [60].
Integration with Lead Optimization Workflow
Homology models gain practical utility when seamlessly integrated into the structure-based lead optimization pipeline. The following workflow demonstrates how computational models inform experimental design in iterative cycles of compound optimization:
Application to Structure-Activity Relationship Analysis
Molecular Docking for Binding Mode Prediction
Molecular docking represents the primary computational method for exploiting homology models in SAR analysis. Docking simulations predict how small molecules interact with the binding site of the target protein, providing atomic-level insights that explain observed structure-activity relationships. The process involves three core components: search algorithms that generate possible binding poses, scoring functions that rank these poses by predicted binding affinity, and validation protocols that ensure predictive reliability [64].
Successful application requires careful workflow validation through docking known active compounds and decoys to establish scoring thresholds and performance metrics. For homology models, particular attention must be paid to binding site flexibility and the potential influence of sequence variations between target and template. Consensus scoring approaches that combine multiple scoring functions often improve correlation with experimental binding data compared to single scoring functions [64].
Quantitative SAR (QSAR) Integration
Homology models enhance traditional QSAR approaches by providing structural context for molecular descriptors. While classical QSAR correlates biological activity with chemical structure using statistical methods, structure-based QSAR incorporates interaction energy terms and spatial relationships derived from the protein-ligand complex [65]. This integrated approach proves particularly valuable during lead optimization when congeneric series exhibit similar binding modes but varying potency due to specific atomic substitutions.
The combination of homology models with molecular dynamics simulations further extends SAR analysis by capturing the dynamic nature of protein-ligand interactions. Short simulations (50-100 ns) can reveal conformational flexibility, binding stability, and the role of water molecules in mediating interactions—all factors that influence structure-activity relationships but remain invisible in static structural models [60].
Experimental Protocols and Validation Strategies
Protocol for Molecular Docking with Homology Models
Objective: To predict binding modes and affinities of lead compounds using a validated homology model for SAR analysis.
Methodology:
- Protein Preparation:
- Import homology model into molecular modeling software (e.g., Schrödinger Maestro, MOE)
- Add hydrogen atoms and optimize protonation states at physiological pH
- Perform constrained energy minimization to relieve steric clashes while preserving overall fold
Binding Site Definition:
- Identify binding pocket through structural alignment with template complex or computational detection methods
- Define binding grid centered on key residues with dimensions sufficient to accommodate ligand flexibility
Ligand Preparation:
- Generate 3D structures from chemical representations
- Assign proper bond orders and ionization states
- Perform conformational sampling to identify low-energy states
Docking Execution:
- Apply flexible ligand docking protocols with appropriate search algorithms
- Generate multiple poses per compound (typically 10-50)
- Score poses using consensus functions that combine empirical, forcefield, and knowledge-based terms
Result Analysis:
- Cluster similar binding poses to identify predominant interaction patterns
- Quantify key interactions (hydrogen bonds, hydrophobic contacts, π-stacking)
- Correlate docking scores with experimental IC₅₀ or Kᵢ values to validate predictive capability [64]
Validation: Redock known crystallographic ligands and compute root-mean-square deviation (RMSD) of predicted versus experimental pose. Successful docking typically achieves RMSD <2.0 Å for the majority of test ligands.
Protocol for Binding Free Energy Calculations
Objective: To quantitatively predict protein-ligand binding affinities for SAR analysis and lead prioritization.
Methodology:
- System Setup:
- Extract protein-ligand complex from docking studies
- Solvate in explicit water model with appropriate counterions
- Apply periodic boundary conditions matching experimental conditions
Equilibration Protocol:
- Perform gradual heating from 0K to 300K over 100ps with positional restraints on protein and ligand
- Conduct equilibrium simulation (1-5ns) without restraints to stabilize system
Production Simulation:
- Run unrestrained molecular dynamics simulation (50-100ns)
- Employ enhanced sampling techniques if necessary (e.g., umbrella sampling, metadynamics)
- Calculate binding free energy using endpoint methods (MM/PBSA, MM/GBSA) or alchemical transformations (TI, FEP)
Data Analysis:
- Compute interaction energies for individual residues to identify binding hotspots
- Decompose free energy contributions by component (electrostatic, van der Waals, solvation)
- Correlate predicted ΔG values with experimental measurements to establish predictive validity [64]
Applications: This protocol enables quantitative comparison of analog series, prediction of affinity for newly designed compounds, and identification of structural modifications that optimize binding interactions.
Research Reagent Solutions
Table 2: Essential Computational Tools for Homology Modeling and SAR Analysis
| Tool Category | Representative Software | Primary Function | Application in SAR |
|---|---|---|---|
| Homology Modeling | MODELLER [60], SWISS-MODEL | Protein structure prediction from sequence | Generate structural models for targets lacking experimental structures |
| Molecular Docking | AutoDock [64], GOLD [64], Glide | Ligand pose prediction and virtual screening | Predict binding modes and rank compound series by affinity |
| Molecular Dynamics | AMBER, GROMACS, Desmond | Simulation of biomolecular motion and interactions | Assess binding stability, conformational changes, and water-mediated interactions |
| Structure Analysis | Pymol, Chimera, MOE | Visualization and analysis of 3D structures | Identify key interactions, map binding sites, and guide molecular design |
| Sequence-Structure Tools | AlphaFold [62] [60], RaptorX [65] | Deep learning-based structure prediction | Model challenging targets with low template identity |
| Free Energy Calculations | Schrödinger FEP+, AMBER TI | Binding affinity prediction from simulations | Quantitatively predict SAR and optimize lead compounds |
Case Studies and Applications
Antibacterial Drug Discovery
Homology modeling has proven particularly valuable in antibacterial drug discovery, where many targets lack experimental structures. For example, homology models of bacterial enzymes such as DNA gyrase and peptide deformylase have successfully guided the optimization of inhibitor potency and selectivity [65]. In one application, researchers developed homology models of AmpC β-lactamase to understand the structural basis of antibiotic resistance and design novel inhibitors that restore the efficacy of β-lactam antibiotics [66].
The integration of homology models with molecular dynamics simulations has provided insights into the conformational flexibility of bacterial targets, revealing allosteric binding sites not apparent in crystal structures. These approaches have accelerated the discovery of novel antibiotic classes addressing multidrug-resistant pathogens, demonstrating the practical impact of computational methods in addressing urgent medical needs [65].
Protein-Protein Interaction Inhibitors
Protein-protein interactions (PPIs) represent challenging targets for drug discovery due to their typically large and shallow interfaces. Homology modeling has facilitated PPI inhibitor development by providing structural models of interaction interfaces that guide the design of stabilizing small molecules or peptide mimetics. For instance, homology models of Bcl-2 family proteins have informed the design of apoptosis-inducing compounds that disrupt pro-survival protein complexes [64].
Recent advances combining homology modeling with deep learning approaches have further improved the accuracy of PPI interface prediction, enabling more reliable drug design for these difficult targets. The RFdiffusion tool has demonstrated particular capability in designing binders for protein targets, generating de novo proteins that interact with specified regions of therapeutic interest [62].
Integration with Artificial Intelligence
The convergence of homology modeling with artificial intelligence represents the most significant advancement in structure-based drug design. AI-driven drug discovery (AIDD) leverages deep learning models trained on the entire Protein Data Bank to predict protein structures and interactions with unprecedented accuracy [61]. AlphaFold 2 and its successor AlphaFold 3 have demonstrated remarkable performance in protein structure prediction, often achieving accuracy comparable to medium-resolution experimental methods [62].
These AI approaches complement rather than replace traditional homology modeling, as evidenced by comparative studies showing that different modeling algorithms excel with different peptide types—AlphaFold and threading complement each other for hydrophobic peptides, while PEP-FOLD and homology modeling show synergy for hydrophilic peptides [60]. The future of homology modeling lies in hybrid approaches that integrate template-based modeling with deep learning predictions, leveraging the strengths of each method while mitigating their individual limitations.
Homology modeling has evolved from a specialized computational technique to an indispensable component of the modern drug discovery toolkit. By providing three-dimensional structural context for biological targets that lack experimental structures, homology models enable rational lead optimization and SAR analysis that would otherwise rely solely on ligand-based approaches. The continued integration of homology modeling with molecular dynamics simulations, free energy calculations, and artificial intelligence promises to further enhance its predictive power and therapeutic impact.
As structural coverage of the proteome expands through initiatives like the AlphaFold Protein Structure Database, and as computational methods continue to advance, homology modeling will play an increasingly central role in drug discovery for challenging targets across therapeutic areas. By adopting the methodologies and best practices outlined in this technical guide, drug discovery researchers can leverage homology modeling to accelerate the development of novel therapeutics addressing unmet medical needs.
Translational Challenges: Optimizing Deep Homology Approaches for Biomedical Research
The paradigm in structural biology has undergone a fundamental shift over the past two decades. Where once existed a substantial "protein structure gap" between the number of known protein sequences and experimentally determined structures, today some form of structural information—either experimental or computational—is available for the majority of amino acids encoded by common model organism genomes [67] [68]. Homology modeling, also known as comparative modeling, has matured into an indispensable tool that bridges this gap by extrapolating from known experimental structures to related sequences [67]. This technical review examines the contemporary landscape of homology modeling, evaluating its successes, limitations, and integration within a broader framework of deep homology in animal design research. We provide researchers and drug development professionals with a critical assessment of methodological approaches, accuracy considerations, and emerging directions in protein structure prediction, with particular emphasis on the sequence-structure-function relationships that underpin evolutionary biology.
The central dogma of structural biology—that a protein's amino acid sequence determines its three-dimensional structure, which in turn dictates its biological function—has guided research for over half a century [69]. Until recently, however, the application of this principle was severely constrained by the "structure knowledge gap" between the exponentially growing number of protein sequences and the relatively small number of experimentally determined structures [67]. The emergence of reliable computational modeling approaches, particularly homology modeling, has fundamentally transformed this landscape.
Homology modeling exploits the observation that evolutionarily related proteins share common structural features, with structural conservation correlating directly with sequence similarity [70]. This approach depends on the robustness of protein structure to changes in primary sequence, whereby certain amino acids experience stronger selective pressure to maintain structural and functional integrity [70]. The method has matured from specialized applications to fully automated pipelines that provide reliable three-dimensional models accessible to non-specialists [67].
Within evolutionary developmental biology ("evo-devo"), the concept of deep homology reveals how conserved genetic toolkits and molecular pathways underlie the development of morphological features across diverse taxa [3]. This framework extends to protein structural biology, where conserved folding principles enable reliable prediction of structure from sequence information across evolutionary distances. The FoxP2 transcription factor network, for instance, exemplifies deep homology in neural circuits supporting learned vocal communication in humans and songbirds [3]. For researchers investigating animal design, homology modeling provides a crucial bridge connecting genomic sequences with structural and functional insights across species.
The Theoretical Basis of Homology Modeling
Fundamental Principles and Assumptions
Homology modeling operates on several foundational principles established through decades of structural comparison:
- Evolutionary Conservation: Protein structure is more conserved than sequence during evolution. While sequences may diverge beyond recognition, the underlying structural scaffold often remains recognizably similar [70].
- Structural Determinism: Similar protein sequences give rise to similar structures and functions, though exceptions exist in regions of the protein universe where similar functions can be achieved by different sequences and structures [69].
- Fold Conservation: The repertoire of protein folds in nature is remarkably limited, with similar structural binding patterns observed across diverse protein-protein interactions [51].
The accuracy of homology modeling critically depends on the sequence identity between the target protein and available templates. Generally, sequence identity above 30% typically yields models with root-mean-square deviation (RMSD) values below 2Å for backbone atoms, while identities in the "twilight zone" (20-30%) present significant challenges and require specialized approaches [71].
The Homology Modeling Workflow
The standard homology modeling protocol consists of several sequential steps, each with distinct methodological considerations:
Figure 1: Homology modeling workflow. The process begins with target sequence analysis and proceeds through template identification, alignment, model building, and rigorous validation.
Template Identification and Sequence Alignment
The initial step involves identifying suitable template structures through sequence similarity searches against structural databases such as the PDB. Sensitive profile-based methods like PSI-BLAST and HMMER have largely replaced basic BLAST for detecting remote homologies [70]. The quality of the target-template alignment constitutes a major bottleneck in generating high-quality models, particularly for sequences with identity below 40% [71].
Advanced alignment methods incorporate structural information to improve accuracy. For example, the ALAdeGAP algorithm implements a position-dependent gap penalty based on solvent accessibility, recognizing that gaps occur more frequently in solvent-exposed regions than in buried residues [71]. This approach significantly improves alignment quality in the twilight zone of sequence identity.
Model Building and Refinement
Once a target-template alignment is established, the actual model construction proceeds through three primary approaches:
- Rigid-body assembly: Copies conserved regions from templates
- Segment matching: Uses overlapping fragments from known structures
- Spatial restraint: Satisfies spatial constraints derived from templates
Model refinement remains a significant challenge, as current methods often fail to consistently improve models closer to the native state [67]. Molecular dynamics simulations can help relax models but require careful parameterization to avoid introducing new errors.
Success Factors and Limitations in Homology Modeling
Key Determinants of Modeling Success
Table 1: Factors influencing homology modeling accuracy
| Factor | High-Accuracy Conditions | Low-Accuracy Conditions |
|---|---|---|
| Sequence Identity | >40% identity to template | <20% identity (twilight zone) |
| Template Quality | High-resolution structure (<2.0Å) | Low-resolution structure (>3.0Å) |
| Alignment Quality | Few gaps, conserved core regions | Many gaps, particularly in secondary structure |
| Domain Architecture | Single domain proteins | Multi-domain proteins with flexible linkers |
| Structural Features | Mainly core secondary structures | Large loops or disordered regions |
The accuracy of homology models depends heavily on template selection and sequence identity. Models based on templates with >50% sequence identity often approach the quality of low-resolution experimental structures [70]. Success rates decline significantly in the "twilight zone" of 20-40% sequence identity, where alignment errors become increasingly common [71].
Multi-domain proteins present particular challenges, as current techniques cannot reliably predict the relative orientation of domains in multi-template models [67]. Additionally, comparative models tend to resemble their templates more closely than the actual target structure, a phenomenon known as "template bias" [67].
Comparative Performance of Modeling Approaches
Table 2: Performance comparison of protein structure prediction methods
| Method | Strengths | Limitations | Best Applications |
|---|---|---|---|
| Homology Modeling | High accuracy with good templates | Template dependence | High-identity targets, conserved folds |
| Threading | Detects distant folds | Limited by fold library | Remote homology detection |
| AlphaFold | High accuracy for monomers | Limited complex accuracy | Monomeric proteins, fold prediction |
| PEP-FOLD | Good for short peptides | Limited to small proteins | Antimicrobial peptides, short segments |
| Ab Initio | Template-free | Computationally intensive | Novel folds without templates |
Recent comparative studies reveal that different modeling algorithms have complementary strengths. AlphaFold and threading approaches complement each other for hydrophobic peptides, while PEP-FOLD and homology modeling show advantages for more hydrophilic peptides [60]. PEP-FOLD provides both compact structures and stable dynamics for most short peptides, while AlphaFold produces compact structures but may not always guarantee stability during molecular dynamics simulations [60].
Advanced Applications and Integrative Approaches
Modeling Protein Complexes and Interactions
While homology modeling has proven highly successful for monomeric proteins, predicting the structures of protein complexes remains challenging. Determining protein complex structures is crucial for understanding cellular processes like signal transduction, transport, and metabolism [51]. Traditional approaches include template-based homology modeling and docking-based prediction methods, but both face limitations [51].
Recent advances in deep learning have significantly improved protein complex structure prediction. DeepSCFold, for instance, uses sequence-based deep learning to predict protein-protein structural similarity and interaction probability, achieving improvements of 11.6% in TM-score compared to AlphaFold-Multimer on CASP15 targets [51]. This method constructs paired multiple sequence alignments (pMSAs) by integrating structural complementarity information, effectively capturing conserved protein-protein interaction patterns [51].
Integrative Structural Biology
With scientific interest moving toward larger macromolecular complexes and dynamic interaction networks, integrative approaches that combine computational modeling with low-resolution experimental data have become essential [67]. These methods can incorporate data from diverse sources including:
- Cryo-electron microscopy (cryo-EM)
- Small-angle X-ray scattering (SAXS)
- Nuclear Magnetic Resonance (NMR)
- Förster resonance energy transfer (FRET)
Integrative modeling allows researchers to study large and complex molecular machines that resist characterization by individual high-resolution methods [67]. The famous Watson-Crick DNA double helix model itself could be considered an early example of integrative modeling, as it was based on low-resolution fiber diffraction data combined with chemical and stoichiometric constraints [67].
Table 3: Key resources for protein structure modeling and analysis
| Resource Category | Examples | Primary Function |
|---|---|---|
| Sequence Databases | UniProt, NCBI | Retrieve target sequences and annotations |
| Structure Databases | PDB, CATH, SCOP | Identify template structures and folds |
| Modeling Servers | SWISS-MODEL, Modeller, HHpred | Automated homology modeling |
| Template Search | HMMER, PSI-BLAST, Jackhmmer | Detect remote homologues |
| Alignment Tools | ALAdeGAP, ClustalOmega, MAFFT | Generate target-template alignments |
| Quality Assessment | MolProbity, PROCHECK, VADAR | Validate model geometry and stereochemistry |
| Specialized Prediction | DeepSCFold, PEP-FOLD, AlphaFold | Complexes, short peptides, general prediction |
Practical Implementation Protocol
For researchers implementing homology modeling, we recommend the following workflow based on current best practices:
Sequence Analysis and Domain Parsing
- Retrieve canonical sequence from UniProt (Swiss-Prot preferred for manual annotation)
- Identify domain boundaries using Pfam, Gene3D, or InterPro
- Parse multi-domain proteins into individual modeling units
Template Identification and Selection
- Perform iterative searches using HMMER or PSI-BLAST against the PDB
- Prioritize templates based on sequence identity, coverage, and resolution
- Consider biological context (bound ligands, oligomeric state, organisms)
Alignment Optimization
- Use profile-based alignment methods
- Incorporate secondary structure prediction to guide gap placement
- Apply structure-aware gap penalties like ALAdeGAP for difficult cases
Model Building and Selection
- Generate multiple models using different protocols
- Assess model quality using multiple metrics (DOPE, GA341, QMEAN)
- Select models with best stereochemistry and packing statistics
Validation and Iteration
- Verify Ramachandran plot statistics, side-chain rotamers, and packing
- Check for structural compatibility with known biological partners
- Iterate through alignment and building steps if necessary
Future Directions and Research Opportunities
The field of computational protein structure prediction continues to evolve rapidly, with several promising research directions emerging:
Addressing Current Limitations
Despite significant progress, important challenges remain in homology modeling. Refinement methods that consistently improve model accuracy toward the native state represent a pressing need [67]. For multi-domain proteins, methods for reliably predicting relative domain orientations and combinations would substantially expand modeling capabilities [67]. Additionally, incorporating protein dynamics and conformational heterogeneity into static structural models remains an important frontier.
Integration with Deep Homology Concepts
The framework of deep homology provides rich opportunities for enhancing protein structure prediction. By analyzing structural conservation across evolutionary distances, researchers can identify core folding principles that transcend sequence similarity [3]. This approach is particularly valuable for modeling proteins from distantly related organisms or evolutionary intermediates.
Recent work on the teleost telencephalon demonstrates how comparative analysis across vertebrate lineages can reveal striking transcriptional similarities between cell-types in fish and subpallial, hippocampal, and cortical cell-types in tetrapods [4]. Such cross-species comparisons provide biological validation for deep homology relationships that can inform structural modeling constraints.
Toward a Complete Protein Structure Universe
Large-scale structure prediction initiatives are progressively saturating the known protein fold space. Analysis of ~200,000 predicted structures from diverse microbial genomes reveals that the structural space is continuous and largely saturated, with the identification of only 148 novel folds beyond those previously characterized [69]. This finding suggests a shift in focus from obtaining structures to contextualizing them within biological systems.
The integration of structural models with functional annotations, as demonstrated by the DeepFRI algorithm, enables residue-specific function prediction across entire proteomes [69]. This approach moves beyond traditional sequence-based annotation to provide mechanistic insights into protein function based on predicted structural features.
Homology modeling has transformed from a specialized computational technique to an essential tool in structural biology, effectively bridging the sequence-structure gap for a majority of proteins in model organisms. Its success, however, remains contingent on template availability and quality, with significant challenges persisting for proteins in the twilight zone of sequence similarity, multi-domain complexes, and conformationally dynamic systems.
The integration of homology modeling with emerging deep learning approaches and experimental data provides a powerful framework for advancing our understanding of protein structure and function. For researchers in evolutionary developmental biology and drug discovery, these methods offer critical insights into the deep homology relationships that shape protein evolution across diverse taxa. As the field continues to mature, the focus is shifting from merely obtaining structural models to effectively interpreting them in their biological context, supporting advanced applications in protein engineering, drug design, and systems biology.
The high rates of attrition in drug development have prompted serious concerns regarding the predictive translatability of animal models to the clinic [72]. Despite significant contributions to our understanding of human health and disease for nearly two thousand years, animal studies inconsistently reproduce the full spectrum of human disease etiology, mechanisms, pathogenesis, and morphology [72]. This translational gap is particularly problematic in pharmaceutical discovery, where animal studies are common in preclinical research for compound evaluation before progression into human clinical trials [72]. To address this challenge, scientists at GlaxoSmithKline (GSK) developed the Animal Model Quality Assessment (AMQA) tool—a structured framework for evaluating animal models to optimize their selection and improve the likelihood of successful clinical translation [72].
The AMQA framework gains additional significance when viewed through the lens of deep homology in animal design research. Evolutionary biology reveals that despite vast morphological differences, deeply conserved genetic and regulatory elements underlie biological systems across species [73] [74]. The discovery of an ancient regulatory syntax (SFZE) controlling brachyury expression across chordates and non-chordates alike demonstrates how conserved genetic programs can be co-opted for novel structures [73]. This evolutionary perspective informs the AMQA's systematic approach to evaluating biological conservation between animal models and humans.
The AMQA Framework: Rationale and Development
The Need for Structured Assessment
The AMQA emerged from an internal after-action review at GSK that examined both successful and unsuccessful clinical outcomes to identify key points of misalignment between preclinical animal pharmacology studies and the clinical trials they supported [72]. This review identified several key features of animal models that contribute to differences in response between animals and human patients, including:
- Fundamental understanding of the human disease of interest
- Biological/physiological context of affected organ systems
- Historical experiences with pharmacologic responses
- How well the model reflects human disease etiology and pathogenesis
- Model replicability and consistency [72]
The framework evolved through multidisciplinary collaboration with in vivo scientists, pathologists, comparative medicine experts, and non-animal modelers, completing three rounds of pilots and iterative design [72]. The challenge was to ensure applicability across a broad portfolio of models for both well-characterized and novel models.
Core Components and Potential Applications
The AMQA tool provides a consistent framework for evaluating animal models to support selection across the spectrum of uses in drug development [72]. Beyond model selection, the AMQA offers multiple additional benefits for the research enterprise:
Table: Potential Applications of the AMQA Framework
| Application | Description | Impact on Research Quality |
|---|---|---|
| Multidisciplinary Partnership | Prompts collaboration between investigators, veterinarians, and pathologists | Enhances study design through diverse expertise |
| Transparent Evaluation | Clearly represents translational strengths/weaknesses of animal models | Supports informed decision-making |
| Knowledge Gap Identification | Highlights areas requiring additional model characterization | Guides targeted research investments |
| Evidence Quality Context | Provides context for data derived from the model | Improves asset progression decisions |
| Harm-Benefit Analysis Support | Enhances ethical review by assessing likelihood of success | Strengthens animal welfare considerations |
The tool's question-based approach makes inputs explicit rather than implicit, focusing on the relevant questions being asked in drug development [72]. This methodology provides a simple yet practical output that clearly identifies strengths and weaknesses of a model, creating a discrete line of sight to the clinical intent that optimizes the likelihood of clinical translation [72].
Implementing the AMQA: Methodological Approach
Assessment Workflow and Process
The AMQA implementation follows a structured workflow that ensures comprehensive evaluation of the animal model for its intended context of use. The assessment process requires multidisciplinary collaboration to properly address all critical aspects of model evaluation.
AMQA Implementation Workflow: The systematic process for conducting an Animal Model Quality Assessment involves multiple stages requiring diverse expertise.
Key Assessment Domains and Scoring Criteria
The AMQA evaluates animal models across several critical domains that collectively determine translational potential. Each domain contributes to an overall assessment of model quality and relevance.
Table: AMQA Scoring Criteria for Model Evaluation
| Assessment Domain | Evaluation Criteria | Scoring Methodology | Weight in Final Assessment |
|---|---|---|---|
| Disease Understanding | Depth of knowledge of human disease pathogenesis and etiology | Qualitative assessment of mechanistic understanding | High |
| Biological Context Conservation | Homology of genetic pathways, systems physiology, and regulatory elements | Quantitative alignment with human biology (e.g., 0-100%) | High |
| Etiological Alignment | Similarity between model induction method and human disease causes | Categorical scoring: High/Medium/Low alignment | Medium |
| Pharmacological Concordance | Historical predictivity of drug responses in the model | Concordance rate with human responses (0-100%) | High |
| Phenotypic Recapitulation | Faithfulness of disease presentation and progression | Multi-parameter scoring of key phenotypes | Medium |
| Replicability & Robustness | Consistency of model performance across laboratories | Statistical measures of variability | Low-Medium |
The scoring system is designed to define predictive translatability, with the completed assessment providing a transparent representation of the model's translational strengths and weaknesses [72]. This enables researchers to understand the limitations of evidence derived from the model and make more informed decisions.
Case Study: AMQA Application to Inflammatory Bowel Disease
Model Evaluation and Assessment
The developers of the AMQA framework provided a comprehensive example of its application to the adoptive T-cell transfer model of colitis as a mouse model mimicking inflammatory bowel disease (IBD) in humans [72]. This case study illustrates how the framework enables systematic evaluation across all critical domains.
In this assessment, researchers evaluated the deep homology between the murine immune system and human immunology, noting strong conservation of T-cell differentiation pathways and inflammatory responses [72] [74]. The model demonstrated high scores for biological context conservation due to these shared immunological mechanisms, though it received moderate scores for etiological alignment since it doesn't fully replicate the complex, multifactorial origin of human IBD [72].
The assessment would have documented the model's strengths in recapitulating specific aspects of human IBD pathology, including T-cell infiltration, epithelial damage, and cytokine profiles, while noting limitations in spontaneous development and complete disease spectrum representation [72]. This nuanced evaluation helps researchers properly contextualize results obtained from this model.
Experimental Protocols for Model Characterization
For researchers implementing the adoptive T-cell transfer colitis model, comprehensive characterization is essential for proper validation. The following protocol details key methodological considerations:
T-cell Isolation and Transfer Protocol:
- Donor T-cell sources: Spleen and lymph nodes from wild-type or genetically modified mice
- CD4+ T-cell enrichment: Magnetic-activated cell sorting (MACS) or fluorescence-activated cell sorting (FACS)
- Cell population selection: Naïve CD4+ CD45RBhigh T-cells for transfer
- Recipient mice: Immunocompromised strains (e.g., Rag1-/- or Rag2-/-)
- Transfer route: Intraperitoneal injection
- Cell dosage: 0.4-0.6 × 10^6 cells per mouse
- Monitoring period: 4-8 weeks post-transfer [72]
Assessment Endpoints:
- Clinical scoring: Weight loss, stool consistency, rectal bleeding
- Pathological evaluation: Colon length, histology scoring of inflammation
- Immune profiling: Cytokine production, T-cell populations in mesenteric lymph nodes
- Molecular analysis: Gene expression of inflammatory mediators [72]
Essential Research Reagents and Materials
Successful implementation of animal models requires specific reagents and materials that ensure consistency and reproducibility. The following table details key solutions for the adoptive T-cell transfer model and general AMQA assessments.
Table: Essential Research Reagent Solutions for Model Implementation
| Reagent/Material | Specification | Research Application | Critical Quality Controls |
|---|---|---|---|
| MACS Separation System | Magnetic cell separation columns and antibodies | Isolation of specific immune cell populations | Antibody specificity, separation efficiency |
| FACS Instrumentation | High-speed cell sorter with 4+ lasers | High-precision cell population isolation | Purity checks, viability assessment |
| Cytokine Profiling Array | Multiplex bead-based immunoassay | Comprehensive inflammatory mediator measurement | Standard curve linearity, detection limits |
| Histopathology Reagents | Tissue fixation, processing, and staining solutions | Morphological assessment of disease pathology | Staining consistency, specificity controls |
| RNA/DNA Isolation Kits | Column-based nucleic acid purification | Molecular analysis of gene expression | Purity measurements (A260/A280 ratios) |
| Next-Generation Sequencing | Library preparation and sequencing reagents | Transcriptomic and genomic characterization | Sequencing depth, quality metrics |
Deep Homology: Evolutionary Perspectives on Model Selection
The concept of deep homology provides a critical evolutionary framework for animal model selection [73]. Deep homology refers to the conservation of genetic regulatory apparatus used to build morphologically distinct features across diverse species [73]. This principle profoundly impacts how researchers should evaluate the translational relevance of animal models.
Conservation of Genetic Regulatory Programs
Research on brachyury gene regulation exemplifies deep homology in animal development. Studies have identified an ancient regulatory syntax (SFZE) consisting of binding sites for four transcription factors in notochord enhancers of chordate brachyury genes [73]. Remarkably, this same regulatory syntax exists in various non-chordate animals and even in Capsaspora, a unicellular relative to animals [73]. These non-chordate SFZE-containing enhancers exhibited activity in the zebrafish notochord, demonstrating the functional conservation of this regulatory code across vast evolutionary distances [73].
This deep homology extends to transposable elements (TEs) that have consistently shaped chromosomal evolution for hundreds of millions of years [74]. Studies in Hydra reveal core sets of a dozen TE elements, mostly DNA transposons, that have been actively maintained across metazoan lineages [74]. These elements contribute to structural variants around loci associated with cell proliferation and long-range topological contacts, influencing genome architecture and regulation across animal species [74].
Implications for Model System Selection
The recognition of deep homology supports more informed selection of model organisms based on conservation of specific biological processes rather than overall morphological similarity. For example, the freshwater cnidarian Hydra provides insights into stem cell dynamics and regenerative processes maintained by three distinct stem cell lineages [74]. Similarly, zebrafish offer advantages for forward genetic screens due to their genetic tractability and conservation of core behavioral components relevant to psychiatric disease [75].
Deep Homology in Model Selection: Conservation of genetic regulatory programs enables informed animal model selection based on shared molecular mechanisms.
Advanced Applications and Future Directions
Integrating Novel Model Systems
The AMQA framework accommodates emerging model systems that leverage evolutionary principles. Cnidarian models like Hydra provide insights into stem cell biology and regeneration, with their three distinct stem cell lineages (endodermal epithelial, ectodermal epithelial, and interstitial/germline stem cells) maintaining distinct identities without interconversion [74]. The telomere-to-telomere genome assemblies of Hydra vulgaris strains reveal how distinct transposable element families are active at both transcriptional and genomic levels via non-random insertions in each lineage [74].
Forward genetic screens in various model organisms continue to identify genes affecting behaviors relevant to human psychiatric disease [75]. These approaches rely on the concept of measurable core component behaviors that contribute to disease phenotypes, such as sensitivity to drug reward, poor impulse control, deficits in pre-pulse inhibition, aggression, disrupted social interaction, anxiety, and sleep disruption [75]. The conservation of fundamental neural processes enables translation of findings from invertebrate to vertebrate models and potentially to humans.
Regulatory Science and 3Rs Integration
The AMQA framework supports ethical research practices and regulatory science. The tool can enhance harm-benefit analysis (HBA) by providing a more rigorous assessment of the "likelihood of success" in ethical review processes [72]. Recent workshops involving the FDA, NIH, and international regulatory bodies have focused on implementing novel methodologies to reduce animal testing while maintaining scientific rigor [76].
In tuberculosis vaccine research, standardized animal models and challenge strains are critical for evaluating protective efficacy [77]. The field is moving toward more diverse model systems, including Collaborative Cross (CC) and Diversity Outbred (DO) mice, which enable researchers to determine correlates of protection and establish vaccine strategies that protect a larger proportion of genetically diverse individuals [77]. These advances align with the AMQA's goal of improving translational predictivity.
The Animal Model Quality Assessment framework represents a systematic approach to one of the most challenging aspects of translational research: selecting appropriate animal models with the highest likelihood of clinical relevance. By providing a structured methodology for evaluating key features of animal models, the AMQA enables more informed model selection, highlights knowledge gaps, and supports better decision-making in drug development.
When integrated with the evolutionary concept of deep homology, the AMQA framework helps researchers leverage conserved genetic and regulatory programs across species while accounting for species-specific differences. This integrated approach advances more biologically informed model selection, potentially improving the translatability of preclinical research and reducing attrition in drug development.
As animal model research continues to evolve, frameworks like the AMQA will be essential for maximizing the scientific value of these critical tools while supporting the ethical principles of the 3Rs (Replacement, Reduction, and Refinement) in biomedical research.
In the field of evolutionary biology and genomics, accurately identifying homologous sequences—genes or proteins sharing common ancestry—is foundational to research on deep homology and animal design. Homology inference, when applied correctly, reveals the remarkable conservation of genetic toolkits and developmental programs across distantly related species. However, a significant challenge persists: distinguishing true evolutionary relationships from random sequence similarities that occur by chance. This guide details the core statistical frameworks, computational protocols, and validation methodologies required to make this critical distinction, thereby ensuring the reliability of subsequent evolutionary and functional analyses.
The principle of homology, defined as "the same organ in different animals under every variety of form and function" [8], is central to comparative biology. With the advent of modern molecular biology, the concept has been extended to the genetic level. The emerging field of deep homology reveals that even morphologically distinct structures in vastly different organisms (e.g., insect and vertebrate limbs) can be regulated by conserved genetic regulatory mechanisms [8]. Establishing true sequence homology is the first and most critical step in uncovering these deep evolutionary connections.
The inference of homology from sequence similarity is based on a simple but powerful logic: when two sequences share more similarity than would be expected by chance, the most parsimonious explanation is common ancestry [39]. The challenge lies in accurately quantifying "expected by chance." This guide provides an in-depth technical overview of the statistical models and experimental protocols designed to meet this challenge, with a focus on applications in evolutionary developmental biology (evo-devo) and drug discovery, where false positives can lead to erroneous conclusions.
Statistical Foundations of Homology Inference
The Extreme Value Distribution for Local Alignment Scores
Sequence similarity search tools like BLAST, FASTA, and SSEARCH use local alignment algorithms. The distribution of scores for local alignments of unrelated sequences follows the Extreme Value Distribution (EVD), not a normal distribution [39] [78].
The probability of observing a local alignment score (S) greater than or equal to a value (x) by chance is given by: [ P(S \geq x) \leq 1 - \exp(-e^{-\lambda x}) ] where (\lambda) is a scaling parameter [39]. In practice, this score is converted into a bit score to normalize for the use of different scoring matrices, which is then used to calculate the final measure of statistical significance [39].
E-values and P-values: Distinguishing Significance
The E-value is the most commonly reported statistic for sequence similarity searches. It represents the expected number of times a given alignment score would occur by chance in a search of a database of a specific size [39].
- Calculation: (E(b) = p(b) \times D), where (p(b)) is the probability of the bit score (b) in a single pairwise comparison, and (D) is the number of sequences in the database [39].
- Interpretation: An E-value of 0.001 means that the observed similarity score is expected to occur by chance once in every 1000 searches of a database of that size. Lower E-values indicate greater statistical significance.
It is crucial to note that E-values are dependent on database size. The same alignment score may be significant in a small database but not in a comprehensive one, not because the homology has changed, but because the multiple testing burden is greater [39]. For DNA:DNA comparisons, which have a shorter "evolutionary look-back time" and less accurate statistics, a much more stringent E-value threshold (e.g., (10^{-10})) is required to infer homology compared to protein-based searches [39].
Table 1: Key Statistical Parameters in Sequence Similarity Searches
| Parameter | Description | Role in Homology Inference |
|---|---|---|
| Raw Score (S) | The numerical score of an alignment based on the chosen substitution matrix and gap penalties. | The initial measure of similarity. Not directly comparable across different searches. |
| Bit Score (b) | A normalized score that accounts for the scoring system used, enabling cross-comparisons. | More usable than the raw score for comparing results across different searches. |
| E-value (E) | The expected number of chance alignments with a score at least as high as the bit score. | Primary metric for statistical significance. Lower E-values indicate greater confidence in homology. |
| P-value | The probability of observing at least one alignment with a given score by chance. | For very small E-values, the P-value and E-value are nearly identical. |
Protocols for Accurate Homology Detection
Core Methodology: Sequence Similarity Searching
The standard workflow for identifying homologs involves querying a sequence against a specialized or comprehensive database.
Experimental Protocol: Protein-Based Homology Search
- Sequence Preparation: Obtain the query protein sequence. If starting with DNA, perform in silico translation to protein, as protein searches are 5-10 times more sensitive due to the higher information content of the 20-letter amino acid alphabet [39].
- Program Selection: Choose a search algorithm.
- BLAST/PSI-BLAST: Fast, heuristic, and widely used. Ideal for initial searches. PSI-BLAST iteratively builds a profile, increasing sensitivity for distant homologs [39].
- SSEARCH/FASTA: Implements the rigorous Smith-Waterman algorithm. Often provides more accurate statistics and is preferred for definitive validation [39] [78].
- HMMER3: Uses profile hidden Markov models, which are highly sensitive for detecting very distant relationships, especially when searching protein domains [39].
- Database Selection: Select an appropriate database (e.g., SwissProt, TrEMBL, NR, or a specialized database for a specific clade).
- Parameter Setting: Use default parameters initially. For distant homology, adjust the E-value threshold to 0.001 or lower. The choice of substitution matrix (e.g., BLOSUM62, BLOSUM45) can be adjusted based on expected divergence.
- Execution and Analysis: Run the search. Collect all hits with an E-value below a predetermined significance threshold (e.g., ( E < 0.001 ) for protein queries) for further validation [39].
Protocol for Validating Suspicious Homology Assignments
When a statistically significant match is scientifically unexpected or could be a potential false positive, additional validation is required [39] [79].
Experimental Protocol: False Positive Detection
- Domain Architecture Check: Examine the domain content of the high-scoring match using tools like InterPro or Pfam. An alignment that links regions with unrelated domains is suspect [39].
- Unrelated Sequence Control: Identify sequences in the database that are unquestionably unrelated (e.g., different structural classes). Check their alignment scores and E-values. If these unrelated sequences also produce significant E-values ((E < 0.01)), the statistical estimates for your query may be inaccurate [39].
- Shuffling-based Significance Estimation: Create shuffled versions of your query sequence that preserve its length and local amino acid composition.
- Tools like SSEARCH can perform this shuffling internally to generate empirical p-values [39].
- Run the search again with the shuffled query against the same database.
- The distribution of scores from these shuffled queries provides a null distribution. If the score of the original alignment falls well within this null distribution, the match is likely a false positive.
- Machine Learning Post-processing: For large-scale genomic or transcriptomic studies, employ a machine learning classifier to identify false positive clusters post-hoc. As demonstrated in one study, such a method successfully reclassified ~25-42% of putative homologies from heuristic algorithms as false positives, particularly in data from low-coverage RNA-seq [79].
The following workflow diagram summarizes the key decision points in the validation process:
The Scientist's Toolkit: Essential Research Reagents and Materials
Table 2: Key Computational Tools and Databases for Homology Research
| Item Name | Type | Primary Function in Homology Research |
|---|---|---|
| BLAST Suite | Software Package | Performs fast, heuristic similarity searches against databases; PSI-BLAST enables more sensitive iterative profile searches [39]. |
| FASTA/SSEARCH | Software Package | Provides rigorous Smith-Waterman alignment; often yields more accurate statistical estimates for gapped alignments [39] [78]. |
| HMMER3 | Software Package | Uses Hidden Markov Models for highly sensitive profile-based searches, ideal for identifying distant homologs and protein domains [39]. |
| SwissProt/UniProt | Protein Database | A high-quality, manually annotated, and non-redundant protein sequence database, essential for reliable initial searches. |
| Pfam/InterPro | Domain Database | Provides domain architecture annotations; critical for validating that a significant alignment involves functionally related units [39]. |
| OrthoDB | Homology Database | A catalog of orthologs; provides pre-computed groups of genes, useful as a reference or for training machine learning models [79]. |
Advanced Considerations in Deep Homology Studies
Research into deep homology often involves comparing sequences from organisms that diverged hundreds of millions or even billions of years ago. At such evolutionary distances, standard sequence-based searches may fail (false negatives), even when the underlying structural and functional relationship is preserved [8].
In these contexts, the concept of homology must be considered at multiple hierarchical levels. While sequences may have diverged beyond recognition, the Gene Regulatory Networks (GRNs) and Character Identity Networks (ChINs) that control development can remain deeply conserved [8]. For instance, the kernel of the GRN specifying heart development is conserved between arthropods and chordates, despite the vast morphological differences in their circulatory organs [8]. Establishing this "deep homology" requires moving beyond simple pairwise sequence comparisons to include:
- Profile and HMM-based Searches: Leveraging information from multiple sequence alignments to build sensitive models of entire protein families.
- Structure Prediction and Comparison: Using homology modeling, which can produce accurate 3D models for proteins even with sequence identities as low as 30%, provided the alignment is accurate [80]. The relationship between sequence identity and structural similarity for membrane proteins is at least as strong as for water-soluble proteins [80].
- Cross-disciplinary Evidence: Integrating gene expression data from RNA-seq with phylogenetic analysis to resolve conflicting hypotheses of character identity, as was done to clarify digit identity in avian wings [8].
Robust statistical validation is the cornerstone of reliable homology inference and, by extension, meaningful research in deep homology and comparative genomics. By understanding and correctly applying the statistical models behind E-values, employing protein-level searches, and utilizing rigorous validation protocols to filter out false positives, researchers can confidently distinguish true evolutionary relationships from misleading chance similarities. As the field moves toward integrating multi-omics data, these foundational practices will remain essential for unraveling the deep historical continuities that shape animal design.
Overcoming Low-Sequence-Identity Hurdles with Profile-Based Methods and Threading
The detection of deep homology through sequence analysis represents a cornerstone of modern computational biology, particularly in animal design research and drug development. When sequence identity falls below the "twilight zone" (typically <25-30%), traditional pairwise alignment methods fail, necessitating advanced profile-based and threading techniques. This technical guide examines the theoretical foundations, practical implementations, and current limitations of these methods, with particular emphasis on their application to short peptide sequences and amyloid signaling motifs relevant to immune response pathways. We present comparative performance data, detailed experimental protocols, and emerging solutions that combine these approaches with deep learning architectures to overcome existing barriers in remote homology detection.
The identification of evolutionarily related proteins with minimal sequence similarity—a phenomenon termed "deep homology"—is crucial for understanding fundamental biological processes in animal development, immune function, and disease mechanisms. In the context of amyloid signaling motifs (ASMs) that facilitate immune response pathways across animals, fungi, and bacteria, this challenge is particularly acute. These short amino acid sequences (approximately 25 residues) adopt cross-β folds capable of self-replication yet exhibit remarkable diversity beyond noticeable homology [81].
The statistical limitations of traditional methods become pronounced when analyzing such sequences. Profile Hidden Markov Models (HMMs), while remaining the standard approach for detecting remote homology for over two decades, suffer from critical weaknesses when applied to short, diverse domains. These models evaluate each alignment position independently (except for indels), lacking the statistical power to capture nonlocal dependencies essential for identifying structurally or functionally similar sequences beyond observable homology [81]. For ASMs and other short but diverse domains, profile HMMs cannot simultaneously maintain both sensitivity and specificity, creating a significant barrier to comprehensive proteome-scale analysis.
Theoretical Foundations and Methodological Approaches
Profile-Based Methods: Extending Beyond Simple Homology
Profile-based methods enhance detection sensitivity by leveraging evolutionary information from multiple sequence alignments (MSAs). The standard profile HMM approach, as implemented in the Pfam database and HMMER software, captures position-specific conservation patterns but operates under the assumption of position independence [81]. This limitation has spurred development of more sophisticated approaches:
Potts Models: These capture pairwise residue-residue correlations in multiple sequence alignments, significantly increasing statistical power. However, they rely on heuristics to avoid combinatorial explosion when calculating sequence-model fit and require alignment, making them unsuitable for modeling functional or structural similarities beyond observable homology [81].
Probabilistic Context-Free Grammatical (PCFG) Models: These flexible models capture nonlocal dependencies at the expense of cubic computational complexity, which remains acceptable for short domains. Currently, PCFG-based models represent the most effective approach for detecting ASMs, outperforming traditional profile methods for certain motif families [81].
Protein Threading: Fold Recognition Beyond Sequence
Threading, or fold recognition, approaches the problem from a structural perspective, aiming to identify the correct structural template for a target sequence from a library of known folds. Despite diverse implementations using different profiles, dynamic programming, hidden Markov models, and pair potentials, best-performing threading methods tend to succeed or fail on the same protein targets [82]. This observation suggests fundamental limitations inherent to the threading approach itself.
Research indicates that threadability depends more on structural features of the target protein than evolutionary distance from templates. Certain folds exhibit high degeneracy, meaning very similar coarse-grained fractions of native contacts can be aligned despite significant structural differences from the native form. For non-threadable proteins, this degeneracy presents an insurmountable barrier [82]. Contemporary threading approaches have consequently reached a performance plateau, with approximately 86% of human proteins having at least one domain predicted with acceptable accuracy (TM-scores ≥ 0.4) but remaining gaps for specific fold classes [82].
Table 1: Quantitative Performance Comparison of Modeling Approaches for Short Peptides
| Modeling Algorithm | Optimal Use Case | Key Strengths | Key Limitations |
|---|---|---|---|
| Profile HMM | Longer sequences with detectable homology | Computational efficiency; well-established benchmarks | Poor performance on short sequences (<40 aa); ignores nonlocal dependencies |
| Threading | Proteins with non-degenerate folds | Leverages structural information directly | Performance plateau; fails on certain fold classes; template-dependent |
| PCFG Models | Short, diverse domains (e.g., ASMs) | Captures nonlocal dependencies; effective for motifs | Cubic computational complexity; less established for proteome-scale |
| AlphaFold | Hydrophobic peptides | High accuracy for many targets; template-free | Limited performance on highly unstable peptides |
| PEP-FOLD | Hydrophilic peptides | De novo approach; compact structures | Variable performance depending on peptide properties |
Integrated and Machine Learning Approaches
Recent advances address limitations of individual methods through integration and machine learning. In comparative studies of computational modeling approaches for evaluating structural dynamics of short-length peptides, researchers have found that AlphaFold and Threading complement each other for more hydrophobic peptides, while PEP-FOLD and Homology Modeling complement each other for more hydrophilic peptides [60]. This suggests that method performance is strongly influenced by peptide physicochemical properties.
Deep learning architectures have shown particular promise for challenging detection tasks. Bidirectional LSTM and BERT-based models trained on diverse motif families and global negative sets can effectively detect amyloid signaling motifs, including novel motifs, even at the genome scale [81]. These approaches demonstrate superior performance compared to grammatical models for certain motif families, suggesting a path forward for proteome-scale detection of remote homologs.
Experimental Protocols and Implementation
Workflow for Remote Homology Detection
The following diagram illustrates a comprehensive workflow for detecting remote homologs using integrated approaches:
Protocol for Comparative Modeling of Short Peptides
For researchers investigating short peptide sequences with potential signaling or antimicrobial functions, the following protocol enables comprehensive structural characterization:
Sequence Acquisition and Preprocessing
- Obtain peptide sequences (typically 12-50 amino acids) from genomic or metagenomic sources
- Use tools like MetaGeneMark for identifying coding regions in metagenomic data
- Translate sequences using EMBOSS Transeq or equivalent
- Filter for appropriate length (e.g., 12-50 amino acids for antimicrobial peptides)
Physicochemical Characterization
- Determine charge using Prot-pi software
- Calculate isoelectric point (pI), aromaticity, grand average of hydropathicity (GRAVY), and instability index using ExPASy-ProtParam
- Predict secondary structure, solvent accessibility, and disordered regions using RaptorX (effective for peptides >26 amino acids)
Multi-Algorithm Structure Modeling
- Apply complementary modeling approaches:
- Homology Modeling (e.g., Modeller): For sequences with detectable templates
- Threading (e.g., PROSPECTOR_4): For fold recognition
- De Novo Methods (e.g., PEP-FOLD3): For template-free modeling
- Deep Learning (e.g., AlphaFold): For end-to-end structure prediction
- Generate multiple models (minimum 4) per sequence using different approaches
- Apply complementary modeling approaches:
Model Validation and Selection
- Perform Ramachandran plot analysis using tools like VADAR
- Conduct molecular dynamics simulations (100ns recommended) to assess stability
- Calculate RMSD, RMSF, and radius of gyration to evaluate structural compactness and dynamics
- Select optimal models based on convergence of results across methods and validation metrics
Table 2: Research Reagent Solutions for Computational Analysis of Remote Homology
| Tool/Category | Specific Implementation | Function/Purpose |
|---|---|---|
| Sequence Analysis | EMBOSS Transeq, MetaGeneMark | Sequence translation and coding region identification |
| Profile Generation | HMMER, HHpred | Build and search with profile HMMs |
| Threading | PROSPECTOR_4, HHpred, SP3 | Fold recognition and alignment |
| Structure Prediction | AlphaFold, PEP-FOLD3, Modeller | 3D structure modeling |
| Specialized Motif Detection | asmscan-bilstm, asmscan-proteinbert | Amyloid signaling motif identification |
| Structure Validation | VADAR, MolProbity | Model quality assessment |
| Dynamics Analysis | GROMACS, AMBER | Molecular dynamics simulations |
| Visualization | Logomaker, PyMOL | Sequence logos and structure visualization |
Case Study: Detection of Amyloid Signaling Motifs
Amyloid signaling motifs present a compelling case study in overcoming low-sequence-identity hurdles. These short sequences (21-40 amino acids) facilitate aggregation into β-sheet-rich structures and function in immune signaling across animals, fungi, and bacteria [81]. Despite their functional conservation, ASMs exhibit extreme sequence diversity with minimal homology, making them ideal test cases for advanced detection methods.
Experimental Framework for ASM Detection
The following protocol outlines a specialized approach for proteome-scale ASM identification:
Dataset Construction
- Curate diverse bacterial and fungal ASM families (e.g., BASS, FASS datasets)
- Include effector-side and receptor-side motifs with varying lengths (11-40 amino acids)
- Create appropriate negative sets representing non-ASM effector domains
- Apply redundancy reduction (e.g., CD-HIT at 70-90% identity)
Model Training and Evaluation
- Implement bidirectional LSTM architectures with embedding layers
- Train BERT-based models on motif families and global negative sets
- Compare against state-of-the-art PCFG models
- Evaluate on remotely related motif families not seen during training
- Test robustness using motif sequences with flanking regions (5-10 amino acid envelopes) and full-length protein termini
Performance Assessment
- Measure sensitivity and specificity across motif families
- Evaluate cross-family generalization capability
- Assess performance on genome-scale datasets
- Compare computational efficiency with traditional methods
Key Findings and Implications
Deep learning approaches demonstrate particular effectiveness for ASM detection, successfully identifying novel motifs even at the proteome scale [81]. The representation learning capabilities of these models enable detection of patterns beyond the scope of homology-based methods, suggesting their utility for other classes of short functional motifs with low sequence conservation.
The relationship between method performance and peptide properties highlights the importance of method selection based on sequence characteristics. This principle extends beyond ASMs to various short peptide families, including antimicrobial peptides and signaling domains.
Future Directions and Integrated Solutions
The emerging consensus points toward integrated approaches that combine the strengths of multiple methodologies. Several promising directions merit attention:
Hybrid Methodologies
- Combine profile information with deep learning representations
- Integrate structural constraints from threading with sequence-based predictions
- Develop meta-predictors that weight methods based on sequence properties
Specialized Architectures
- Design domain-specific models for particular protein families
- Develop attention mechanisms that explicitly capture long-range dependencies
- Create interpretable models that provide biological insights beyond predictions
Scalable Implementations
- Optimize algorithms for proteome-scale analysis
- Develop efficient screening pipelines for large sequence databases
- Create standardized benchmarks for remote homology detection
The integration of these approaches promises to extend our ability to detect deep homology relationships, with significant implications for understanding animal design principles, evolutionary mechanisms, and developing novel therapeutic strategies.
Integrating Multi-Model Strategies to Mitigate Biological Differences and Improve Predictivity
The principle of deep homology provides a critical conceptual framework for understanding how disparate species and biological systems, from arthropods to chordates, utilize a remarkably conserved toolkit of genetic regulatory mechanisms to build morphologically diverse structures [8]. This discovery, propelled by modern evolutionary developmental biology (evo-devo), suggests that anatomical features not considered homologous by strict historical definitions can be regulated by the same, deeply conserved genetic circuits [8] [83]. For instance, the specification of heart development in clades as distant as arthropods and chordates relies on a core set of regulatory interactions, tracing back to a primitive circulatory organ at the base of the Bilateria [8].
In the age of high-throughput biotechnology, this concept extends beyond morphology to the realm of data. Just as deep homology reveals a shared regulatory logic underlying apparent diversity, multi-modal data integration seeks to uncover the shared biological logic underlying the complex, high-dimensional data measured from cells and tissues. Current technologies can simultaneously measure multiple modalities—such as RNA, DNA accessibility, and protein—from the same cells [84]. However, the analytical challenge mirrors the biological one: to move beyond a partial picture and integrate these disparate data types into a unified understanding of cellular function and regulation [84]. This paper explores how multi-model computational strategies are essential for mitigating biological differences, or "noise," inherent in single-modality analyses, thereby improving the predictive power of models in biomedicine and facilitating discoveries within a framework inspired by deep homology.
Multi-Modal Data Types and Computational Challenges
The first step in a multi-modal analysis is understanding the nature and source of the data. Different modalities provide complementary views of the biological state, and their successful integration hinges on recognizing their unique characteristics.
Table 1: Primary Data Types in Multi-Modal Biological Analysis
| Data Class | Example Measurements | Key Technologies | Biological Insight Provided |
|---|---|---|---|
| Genetic Data | Single Nucleotide Polymorphisms (SNPs), Copy Number Variations (CNVs) [85] | Whole-exome sequencing, Whole-genome sequencing (WGBS) [85] | DNA sequence variation linked to disease predisposition [85]. |
| Proteogenomic Data | mRNA, micro RNA (miRNA) expression [85] | RNA-seq [85] | Gene expression activity and transcriptome-wide regulation [85]. |
| Proteomic Data | Protein abundance and synthesis dynamics [85] | Mass Spectrometry (MassSpec) [85] | Functional gene product levels and cellular signaling activity. |
| Epigenomic Data | DNA methylation (DNAm), Chromatin accessibility, Histone modifications [85] | ATAC-seq, ChIP-seq [85] | Regulatory landscape and function of genomic regions without changing DNA sequence. |
| Imaging Data | Whole Slide Images (WSIs) of tissues, Magnetic Resonance Imaging (MRI) [85] | Slide scanners, MRI machines [85] | Cellular and anatomical morphology, disease pathology, and spatial context. |
| Clinical Data | Blood pressure, inflammatory markers, age, sex, drug history [85] | Medical examinations & records [85] | Patient phenotype, treatment history, and overall health status. |
The integration of these data types is non-trivial and presents several distinct computational challenges [85]:
- Missing Values: Very common in omics data due to technical dropouts or limited sensitivity of instruments.
- Dimensionality Mismatch: Integrating low-dimensional clinical data with high-dimensional omics data is challenging for standard algorithms.
- Interpretability: A trade-off exists where models with the highest predictive power, particularly deep-learning models, are often "black-box" in nature.
- Incorporating Prior Knowledge: Effectively integrating existing biological knowledge (e.g., gene sets, protein-protein interactions) into models to boost performance and aid interpretability is a complex task.
A common strategy to address these challenges is representation learning, where unobservable latent variables are inferred from the observed high-dimensional data. In multimodal representation learning, a joint representation across all modalities is inferred to paint a comprehensive picture of the underlying biological processes [85].
Methodological Approaches: From Multi-Omics to Multi-Modal Integration
Computational methods for multi-modal data integration have evolved to address the four key predictive tasks in biomedicine: patient survival prediction, disease biomarker and subtype classification, therapy response prediction, and clinical decision making [85]. These methods can be broadly categorized, with some specializing in multi-omics data and others, like UnitedNet, offering a more generalizable framework.
The UnitedNet Framework: An Explainable Multi-Task Model
UnitedNet is an explainable multi-task deep neural network designed as a comprehensive end-to-end framework for single-cell multi-modality data analysis [84]. Its architecture is specifically engineered to perform both joint group identification (e.g., cell type identification) and cross-modal prediction (e.g., predicting DNA accessibility from RNA expression) within a unified model, which has been shown to improve performance on both tasks compared to single-task models [84].
Experimental Protocol and Workflow: The UnitedNet model employs an encoder-decoder-discriminator structure that does not presume known data distributions, allowing it to handle the heterogeneous statistical characteristics of different modalities [84]. The training process involves an overarching loss function that combines:
- A clustering or classification loss for group identification.
- A contrastive loss to align latent codes from the same cell and separate codes from different cells.
- A reconstruction loss to ensure latent codes accurately represent the original data.
- A prediction loss and an adversarial loss (via a discriminator) to improve the accuracy of cross-modal predictions [84].
During training, the network parameters are optimized by alternately training between the joint group identification and cross-modal prediction tasks, which are linked through a shared latent space [84]. This multi-task learning approach reinforces the shared latent representations, as demonstrated on a simulated four-modality Dyngen dataset, where multi-task learning led to more separable and distinct latent clusters compared to single-task learning [84].
Diagram 1: UnitedNet's Multi-Task Learning Workflow. The architecture shows how multi-modal data is encoded into a shared latent space, which is then used for two alternating tasks: joint group identification and cross-modal prediction.
A Landscape of Multi-Modal Methods
UnitedNet is part of a wider ecosystem of methods. The table below summarizes other notable approaches, highlighting the diversity of techniques being applied.
Table 2: Selected Multimodal Data Integration Methods in Predictive Biomedicine
| Model Name | Core Methodology | Primary Data Types | Key Predictive Task(s) |
|---|---|---|---|
| MOFA [85] | Latent Variable Model (LVM) | Multi-omics | Disease subtype classification |
| MOGONET [85] | Graph Convolutional Network (GCN) | Multi-omics | Disease biomarker classification |
| SALMON [85] | Deep Neural Network (DNN) | Multi-omics | Patient survival prediction |
| MCAT [85] | Multimodal Co-Attention Transformer | Genomics, WSIs | Survival prediction, therapy response |
| UnitedNet [84] | Multi-task DNN (Encoder-Decoder-Discriminator) | Single-cell multi-omics, Multi-sensing, Spatial-omics | Joint group identification, Cross-modal prediction |
Experimental Validation and Benchmarking
Robust benchmarking is essential to validate the performance of any new multi-modal method. UnitedNet was rigorously tested on a variety of datasets, including simulated data with ground truth and real-world multi-modality data from Patch-seq, multiome ATAC + gene expression, and spatial transcriptomics [84].
Experimental Protocol for Benchmarking:
- Dataset: A simulated four-modality (DNA, pre-mRNA, mRNA, protein) dataset was generated using Dyngen, a biological process simulator [84].
- Baseline Comparisons: UnitedNet was benchmarked against state-of-the-art multi-modal integration methods including Schema, Multi-Omic Factor Analysis (MOFA), totalVI, and Weighted Nearest Neighbor (WNN) [84]. Single-modality Leiden clustering was used as a performance baseline.
- Evaluation Metric: For unsupervised joint group identification, the integrated joint representations from each method were clustered using the Leiden algorithm [84]. The accuracy of the cluster labels against the ground truth was then measured.
- Ablation Study: To test the necessity of its core components, UnitedNet was also evaluated without its multi-task learning scheme (i.e., without cross-modal prediction) and without its adversarial discriminator (termed a dual-autoencoder) [84].
Quantitative Results: The benchmark results demonstrated that UnitedNet consistently achieves similar or better unsupervised joint group identification accuracy compared to both baseline methods and other state-of-the-art methods [84]. Crucially, the ablation analysis confirmed the importance of the multi-task learning framework, as the group identification accuracy decreased when the cross-modal prediction task was removed [84]. Similarly, the average cross-modal prediction accuracy was reduced in the absence of the adversarial discriminator [84]. This provides strong evidence that the integrated multi-task approach and the encoder-decoder-discriminator structure are key to its robust performance.
Successful multi-modal analysis often relies on leveraging large, publicly available datasets and specific computational tools. The following table details key resources for acquiring data and the core components of a method like UnitedNet.
Table 3: Research Reagent Solutions for Multi-Modal Analysis
| Resource / Component | Type | Function and Utility |
|---|---|---|
| TCGA (The Cancer Genome Atlas) [85] | Data Resource | Provides a comprehensive collection of multi-omic, image, and clinical data for multiple cancer types, serving as a primary benchmark dataset. |
| GTEx (Genotype-Tissue Expression) [85] | Data Resource | Offers genetic and RNA expression data from diverse normal human tissues, useful for understanding baseline biological variation. |
| UK Biobank [85] | Data Resource | One of the largest datasets linking genetic data with extensive imaging and clinical data from participants, enabling population-scale studies. |
| SHAP (SHapley Additive exPlanations) [84] | Computational Tool | An explainable AI algorithm used to dissect trained models like UnitedNet to quantify cell-type-specific, cross-modal feature relevance. |
| Adversarial Discriminator [84] | Model Component | A network that competes with the generator (decoder) to improve the realism and accuracy of cross-modal predictions. |
| Contrastive Loss Function [84] | Model Component | A training objective that maximizes agreement between modality-specific latent codes from the same cell while minimizing agreement with codes from other cells. |
Interpretation and Biological Insight: The Path to Predictivity
The ultimate goal of these methods is to derive biologically and clinically actionable insights. A key advantage of explainable models like UnitedNet is their ability to move beyond prediction to discovery. By applying post hoc explainable machine learning methods, such as the SHAP algorithm, to a trained UnitedNet, researchers can directly quantify the cell-type-specific, cross-modal feature-to-feature relevance [84]. This means the model can not only predict, for example, DNA accessibility from gene expression but can also identify which specific genes are most important for regulating which specific DNA accessible sites in a particular cell type.
This capability aligns with the deep homology concept by providing a computational means to uncover the "conserved regulatory kernels" or "character identity networks" (ChINs) that operate across different modalities and potentially across species [8]. For instance, a multi-modal analysis could reveal that the development of a novel morphological structure in one species relies on a ChIN that is deeply homologous to a network governing a different structure in a distantly related species, all by analyzing the shared regulatory logic in their respective multi-omics data.
Diagram 2: From Model to Biological Insight. The workflow for interpreting a trained multi-modal model to generate testable biological hypotheses about gene regulation.
The integration of multi-model computational strategies represents a paradigm shift in biological research and predictive biomedicine. By simultaneously analyzing multiple data modalities within unified frameworks like UnitedNet, researchers can mitigate the limitations and biological noise inherent in single-modality analyses. This leads to more accurate cell type identification, more robust cross-modal predictions, and, critically, the ability to discover and quantify the regulatory relationships that define cellular identity and function. This approach, framed by the evolutionary concept of deep homology, provides a powerful lens through which to interpret complex biological systems. It suggests that just as a conserved genetic toolkit underlies morphological diversity, a conserved data-analytic toolkit can be developed to uncover the unifying principles hidden within multi-modal biological data, ultimately enhancing our ability to predict disease, drug response, and patient outcomes.
Evidence and Efficacy: Validating Deep Homology in Disease Modeling and Therapeutic Development
Protein homolog detection serves as a fundamental pillar in computational biology, essential for virtually all biological sequence-related research, including protein structure prediction, biomolecular functional analysis, transcription regulation studies, novel enzyme discovery, and phylogenetic reconstruction [86]. Within the context of animal design research, the precise identification of homologous proteins—evolutionarily related sequences with similar structures and functions—enables scientists to trace deep homological relationships across diverse species. These relationships reveal how conserved genetic blueprints have been modified through evolutionary processes to generate the remarkable diversity of animal forms and functions observed in nature [4]. The detection of remote homologs, which share common ancestry but have diverged significantly in sequence, presents particular challenges for traditional alignment-based methods, often causing them to miss critical evolutionary connections that underlie deep homology principles.
The rapid expansion of protein sequence databases, driven by next-generation sequencing technologies, has created an urgent need for more sensitive and computationally efficient homolog detection methods [87]. This whitepaper provides a comprehensive technical benchmarking of three distinct approaches to protein homolog detection: the novel Dense Homolog Retriever (DHR) framework, the established profile-based PSI-BLAST method, and the hidden Markov model-based HMMER toolset. By evaluating their relative performance in terms of sensitivity and speed, we aim to provide researchers with actionable insights for selecting appropriate tools based on their specific research requirements, particularly those investigating deep homological relationships in animal systems.
DHR: Deep Learning-Based Dense Retrieval
DHR represents a paradigm shift in protein homolog detection, moving from traditional sequence alignment to an embedding-based retrieval system. Its core innovation lies in encoding protein sequences into dense embedding vectors using a protein language model initialized with Evolutionary Scale Modeling (ESM) [86] [88]. The system employs a dual-encoder architecture that generates different embeddings for the same protein sequence depending on its role as a query or candidate sequence, providing greater flexibility in homology matching [86]. Through contrastive learning techniques, the model learns to embed positive input pairs nearby in the embedding space while pushing negative pairs apart, enabling the capture of rich coevolutionary and structural information without explicit alignment [86].
The alignment-free nature of DHR eliminates the computational overhead associated with progressive alignment or dynamic programming used in conventional methods [86]. After the training phase, the system generates offline protein sequence embeddings of high quality, then leverages these embeddings and similarity search algorithms to retrieve homologs for each query protein. By designating embedding similarity as the retrieval metric, DHR can identify structurally similar proteins more accurately than traditional methods, particularly for remote homologs with low sequence similarity [88].
PSI-BLAST: Position-Specific Iterated BLAST
PSI-BLAST (Position-Specific Iterated Basic Local Alignment Search Tool) represents an enhancement of the traditional BLAST algorithm, employing an iterative approach to build position-specific scoring matrices (PSSMs) that capture conserved patterns in protein families [86]. The method begins with a standard BLAST search, then constructs a multiple sequence alignment from significant hits to build a PSSM, which is used in subsequent search iterations. This iterative profile-building process improves sensitivity for detecting distant relationships compared to single-pass BLAST searches [86].
Despite its improvements over BLAST, PSI-BLAST still faces limitations in detecting remote homologs with very low sequence similarity, as it remains fundamentally dependent on sequence alignment and can miss structural relationships not evident from primary sequence alone [86]. The method also requires multiple database passes, increasing computational time compared to single-pass methods.
HMMER: Hidden Markov Model-Based Detection
HMMER applies hidden Markov models (HMMs) to protein homolog detection, using probabilistic models that capture position-specific information about conserved domains [89] [90]. The toolset includes various implementations, with HMMER2 operating in "glocal" mode (enforcing full domain-to-sequence alignments) and HMMER3 utilizing local alignment mode for massive speed improvements [89]. The glocal alignment mode of HMMER2 is particularly valued for precise domain annotation, as it ensures complete alignments to domain models, with each domain representing a unit of function [89].
HMMER3's quantum leap in computation speed makes large-scale domain annotation practical on standard computers, but it cannot reproduce HMMER2's glocal mode alignments, working instead in fragmented domain alignment mode [89]. This limitation has prompted hybrid approaches like xHMMER3x2, which combines HMMER3's speed with HMMER2's alignment completeness for improved large-scale protein domain annotation [89] [90]. HMMER's effectiveness is highly dependent on the quality of the profile, which can only be obtained from multiple sequence alignments that are not always available [86].
Experimental Design and Benchmarking Methodology
Datasets and Evaluation Metrics
To ensure fair and comprehensive benchmarking, the evaluation of DHR against traditional methods utilized the structural classification of proteins (SCOPe) database, a carefully curated repository of protein structural domains organized in a hierarchy based on structural and evolutionary relationships [86]. SCOPe provides a reliable ground truth for assessing detection sensitivity across different levels of evolutionary relationships, from close family members to distant superfamily relations.
For large-scale performance validation, studies also incorporated the BFD/MGnify dataset, a massive database of approximately 300 million protein sequences that enables exploration of a wider spectrum of protein diversity [88]. Training sets included 2 million query sequences selectively chosen from UR90, with JackHMMER used to iteratively search for candidate sequences in Uni-Clust30 [88].
The primary evaluation metrics included:
- Sensitivity: Measured as the ability to detect true homologs across different levels of the SCOPe hierarchy (family, superfamily) [86]
- Area Under the Curve before First False Positive (AUC-1FP): A stringent metric evaluating the trade-off between sensitivity and specificity [88]
- Execution Time: Comparing the computational efficiency across methods for database searching [86]
- Multiple Sequence Alignment (MSA) Quality: Assessed through the effective number of sequences (Meff) and impact on downstream applications like protein structure prediction [86]
Experimental Protocols
Homolog Detection Protocol:
- For each method, query sequences were selected from SCOPe database
- Searches were conducted against reference databases (UniRef90 for standard tests, BFD/MGnify for large-scale tests)
- Significant hits were validated against known structural classifications in SCOPe
- True positives, false positives, and false negatives were calculated at family and superfamily levels
- Execution times were measured using consistent hardware specifications
MSA Construction and Evaluation Protocol:
- Homologs identified by each method were processed with JackHMMER to construct MSAs
- MSA quality was assessed by diversity (Meff) and number of sequences
- MSAs were fed to AlphaFold2 for structure prediction
- Prediction accuracy was measured by root-mean-square deviation (r.m.s.d.) against experimental structures
- Performance was compared across CASP13 domain (CASP13DM) and CASP14 domain (CASP14DM) targets
Table 1: Key Research Reagents and Computational Resources
| Resource Name | Type | Purpose in Benchmarking | Key Characteristics |
|---|---|---|---|
| SCOPe Database [86] | Structural Database | Ground truth for homolog validation | Curated hierarchy of structural domains |
| BFD/MGnify [88] | Sequence Database | Large-scale performance testing | ~300 million protein sequences |
| UniRef90 [88] | Sequence Database | Standardized testing environment | Clustered sequences at 90% identity |
| JackHMMER [86] | Software Tool | MSA construction from homologs | Iterative search algorithm |
| ESM (Evolutionary Scale Modeling) [86] | Protein Language Model | Base model for DHR embeddings | Pre-trained on millions of sequences |
Quantitative Performance Comparison
Sensitivity Analysis
The benchmarking results demonstrate that DHR achieves significantly higher sensitivity compared to traditional methods, particularly for challenging remote homolog detection. Quantitative analysis reveals a >10% increase in overall sensitivity compared to previous methods and a remarkable >56% increase in sensitivity at the superfamily level for samples that are difficult to identify using alignment-based approaches [86] [87]. When evaluated on the SCOPe dataset, DHR maintained 100% sensitivity for many queries while other methods showed substantial degradation, especially at the superfamily level [88].
In a specific case study involving the d1w0ha query, neither PSI-BLAST nor MMseqs2 matched any results, while DHR successfully retrieved five homologs that were classified as the same family as d1w0ha in SCOPe [88]. Overall, DHR detected the most homologs with a sensitivity of 93% in this evaluation, demonstrating its superior ability to integrate structural information for homolog detection [88].
The AUC-1FP metric, which measures the area under the curve before the first false positive, further confirmed DHR's advantage with a score of 89%, significantly outperforming other methods [88]. At the more challenging superfamily level, where all methods experienced approximately 10% performance degradation, DHR maintained its leading position with an AUC-1FP score of 80% [88].
Table 2: Sensitivity Comparison Across Homolog Detection Methods
| Method | Overall Sensitivity | Superfamily-Level Sensitivity | AUC-1FP Score | Specialization |
|---|---|---|---|---|
| DHR [86] [88] | >10% increase vs. other methods | >56% increase for challenging cases | 89% (Overall), 80% (Superfamily) | Remote homologs, structural detection |
| PSI-BLAST [86] | Baseline | Significant degradation at superfamily level | Lower than DHR | Family-level detection |
| MMseqs2 [86] | Moderate | Moderate degradation at superfamily level | Lower than DHR | Balanced performance |
| HMMER3 [89] [90] | ~95.7% (for domain annotation) | Varies by model and alignment mode | N/A | Domain annotation, glocal alignment |
| DIAMOND [86] | Moderate | Limited for remote homologs | Lower than DHR | Fast alignment |
Speed and Efficiency Benchmarks
The computational efficiency of DHR represents one of its most significant advantages over traditional methods. Comprehensive benchmarking demonstrates that DHR is up to 22 times faster than traditional methods such as PSI-BLAST and DIAMOND, and up to 28,700 times faster than HMMER [86] [87]. This dramatic speed improvement enables researchers to search a dataset of 70 million entries in just a few seconds on a single graphics processing unit (GPU), with linear scaling as database size increases [86].
When incorporated into MSA construction pipelines, DHR with JackHMMER proved to be 93 times faster than default JackHMMER approaches while constructing highly consistent MSAs with AlphaFold2's default MSAs on CASP13 and CASP14 datasets [86]. Furthermore, DHR can construct the same number of homologs of different lengths in constant time, while JackHMMER's performance scales linearly with sequence length and database size [88].
Table 3: Computational Efficiency Comparison
| Method | Speed Relative to DHR | Hardware Requirements | Database Scaling | Typical Use Case |
|---|---|---|---|---|
| DHR [86] [87] | Baseline (22x faster than PSI-BLAST) | Single GPU | Linear | Large-scale database searching |
| PSI-BLAST [86] | 22x slower than DHR | CPU | Multiple iterations required | Medium-sized databases |
| DIAMOND [86] | 22x slower than DHR | CPU | Linear | Fast alignment searches |
| HMMER [86] | 28,700x slower than DHR | CPU | Dependent on profile quality | Precise domain annotation |
| JackHMMER [86] | 93x slower than DHR+JackHMMER | CPU | Linear with sequence length | Iterative profile building |
Implementation Workflows and Integration
DHR Implementation Architecture
DHR System Architecture and Workflow
The DHR implementation follows a streamlined workflow that begins with protein sequence input processed through the ESM protein language model to generate initial representations [86]. The dual-encoder architecture then transforms these representations into specialized embeddings for query and database sequences, optimized through contrastive learning to ensure that homologous pairs are embedded nearby in the vector space while non-homologous pairs are separated [86]. The resulting embeddings are stored for efficient retrieval, enabling rapid similarity searches against large databases without recomputation.
For integration with existing structural bioinformatics pipelines, DHR outputs identified homologs to JackHMMER for MSA construction, which can then be directly fed to AlphaFold2 for protein structure prediction [86]. The hybrid DHR-meta approach, which combines DHR and AlphaFold2 default MSAs, has demonstrated superior performance on CASP13DM and CASP14DM targets, outperforming individual pipelines [86].
Traditional Method Workflows
Traditional Method Workflows: PSI-BLAST and HMMER
PSI-BLAST operates through an iterative process beginning with an initial BLAST search against the target database [86]. Significant hits from this search are used to construct a position-specific scoring matrix (PSSM) that captures conserved patterns in the protein family. This PSSM is then used for subsequent search iterations, with the process repeating until convergence is achieved (no new significant hits are found) or a predetermined number of iterations is completed [86].
HMMER implementations follow a different approach, beginning with the construction of a hidden Markov model profile either from existing multiple sequence alignments or through iterative search procedures like JackHMMER [89]. This profile is then used to search sequence databases, with HMMER3 utilizing local alignment mode for speed and HMMER2 employing glocal alignment mode for more complete domain-to-sequence alignments [89]. The xHMMER3x2 framework represents a hybrid approach that uses HMMER3 for initial domain detection followed by HMMER2 for glocal-mode sequence-to-full-domain alignments, balancing speed and alignment completeness [89].
Implications for Deep Homology Research in Animal Systems
The enhanced sensitivity and speed of DHR for detecting remote homologs has profound implications for research in deep homology—the principle that shared genetic regulatory circuits underlie the development of analogous structures across diverse animal lineages [4]. Studies of the teleost telencephalon have revealed striking transcriptional similarities between cell-types in fish and subpallial, hippocampal, and cortical cell-types in tetrapods, supporting partial eversion of the teleost telencephalon and demonstrating deep homology in vertebrate forebrain evolution [4]. The ability to detect such distant relationships depends critically on sensitive protein homolog detection methods.
In practical applications, DHR's capacity to identify remote homologs enables researchers to:
- Trace Evolutionary Trajectories: Follow the modification of protein families across diverse animal taxa to understand how ancestral genetic toolkits have been adapted for specialized functions [4]
- Identify Conserved Regulatory Elements: Detect distantly related transcription factors and signaling molecules that regulate development in animal systems [4]
- Bridge Model Organisms: Connect research findings from genetically tractable model organisms (like zebrafish and Drosophila) to other species, including humans [4]
- Annotate Novel Genomes: Rapidly characterize protein coding genes in newly sequenced genomes by transferring functional annotations from established model systems [89]
The computational efficiency of DHR makes large-scale comparative genomics approaches feasible, enabling researchers to perform systematic homolog detection across multiple genomes to identify deeply conserved genes and regulatory pathways that define animal body plans and physiological systems.
Comprehensive benchmarking demonstrates that DHR represents a significant advancement in protein homolog detection, offering substantially improved sensitivity for remote homolog detection while dramatically reducing computational requirements compared to traditional methods like PSI-BLAST and HMMER. These improvements directly benefit research in deep homology and animal design by enabling more comprehensive detection of evolutionarily conserved elements across diverse species.
Future developments in protein language models and retrieval techniques promise further enhancements to homolog detection sensitivity and efficiency. The integration of structural information directly into embedding models, multi-modal approaches combining sequence and structural features, and specialized models for particular protein families represent promising research directions. As these methods mature, they will further accelerate our understanding of how evolutionary processes have generated animal diversity through the modification of shared genetic blueprints.
For researchers investigating deep homology in animal systems, we recommend DHR for initial large-scale database searches and remote homolog detection, while traditional methods like HMMER with glocal alignment remain valuable for precise domain annotation tasks. The integration of these complementary approaches through hybrid frameworks will provide the most comprehensive insights into protein evolution and function.
The journey from preclinical discovery to clinical success represents one of the most significant challenges in modern biomedical research. In both inflammatory bowel disease (IBD) and oncology, the translation of basic scientific findings into effective clinical therapies has been hampered by biological complexity and model system limitations. The overall rate of successful translation from animal models to clinical cancer trials is less than 8%, highlighting the critical need for more predictive approaches [91]. This translational failure rate originates from fundamental disparities between model systems and human pathophysiology, including genetic, molecular, immunologic, and cellular differences that prevent animal models from serving as fully effective predictors of human outcomes [91].
Within this challenging landscape, the evolutionary developmental biology (evo-devo) concept of "deep homology" provides a valuable conceptual framework. Deep homology describes the phenomenon whereby disparate aspects of morphology—often in distantly related organisms—are regulated by the same conserved genetic regulatory mechanisms [8] [83]. These deeply conserved genetic circuits represent a paradox: while the anatomical structures themselves may not be homologous in a classical sense, the underlying developmental mechanisms exhibit profound evolutionary conservation. This principle extends to disease mechanisms, suggesting that conserved biological pathways may offer superior targets for therapeutic intervention compared to species-specific physiological responses [8] [21].
Table 1: Key Challenges in Clinical Translation Across Therapeutic Areas
| Challenge Area | IBD Context | Cancer Context |
|---|---|---|
| Model Limitations | Immunosuppressive therapies tested in models not accounting for disease heterogeneity [92] | Overreliance on rodent models that overestimate efficacy by ~30% [91] |
| Success Rates | Complete remission achieved in only ~30% of patients with current therapies [92] | Less than 8% of drugs passing successfully from animal models to Phase I trials [91] |
| Biological Complexity | IBD represents a continuum of disorders with >240 genetic loci identified [92] [93] | Crucial transcription factor binding sites differ between humans and mice in 41-89% of cases [91] |
| Predictive Biomarkers | Lack of validated predictive biomarkers for treatment selection [92] [94] | Fewer than 1% of published cancer biomarkers enter clinical practice [95] |
Deep Homology: A Conceptual Framework for Translational Research
Historical Development and Core Principles
The concept of deep homology emerged from comparative evolutionary developmental biology (evo-devo) as researchers discovered that distantly related species utilize remarkably conserved genetic toolkits during embryogenesis. The term was originally coined to describe the repeated use of highly conserved genetic circuits in the development of anatomical features that do not share homology in a strict historical or developmental sense [8]. For example, despite evolutionary separation since the Cambrian period, the development of insect and vertebrate appendages shares striking similarities in the specification of their embryonic axes, all regulated by homologous genetic pathways [8].
This conceptual framework has evolved through several related formulations that emphasize different aspects of regulatory conservation:
- Kernels: These represent sub-units of gene regulatory networks (GRN) that are central to bodyplan patterning, exhibit deep evolutionary conservation, and are refractory to regulatory rewiring. Their static behavior underlies the stability exhibited by different animal bodyplans [8].
- Character Identity Networks (ChINs): These are gene regulatory networks that define specific morphological characters and are repeatedly re-deployed during embryogenesis across generations [8].
- Deep Homology: The original concept emphasizes that the same conserved genetic machinery can be deployed in the development of non-homologous structures in distantly related species [83] [21].
Relevance to Disease Modeling and Therapeutic Development
The deep homology framework provides crucial insights for translational medicine by highlighting that conserved genetic circuitry may offer more reliable therapeutic targets than species-specific physiological responses. When developmental signaling pathways such as Notch, Ras/MAPK, Hedgehog, Wnt, TGFβ, and JAK/STAT are conserved across evolution, they likely represent fundamental biological processes with minimal redundancy [21]. This evolutionary conservation suggests that interventions targeting these core pathways may demonstrate more consistent effects across model systems and human patients.
Table 2: Conserved Developmental Signaling Pathways with Translational Relevance
| Pathway | Evolutionary Conservation | Disease Relevance | Therapeutic Targeting Examples |
|---|---|---|---|
| Hedgehog | Conserved from insects to humans [8] | Chondrosarcoma, other solid tumors [91] | IPI-926 (saridegib) - failed in Phase II despite animal success [91] |
| Notch | Widely conserved throughout animal kingdom [21] | Multiple sclerosis, rheumatoid arthritis, cancers [91] | TGN1412 - catastrophic failure in clinical trials despite animal safety [91] |
| Wnt | Conserved across bilaterians [8] | Colorectal cancer, tissue regeneration | Multiple candidates in development |
| Matrix Metalloproteinases (MMPs) | Conserved enzymatic functions | Cancer invasion, metastasis [91] | Multiple MMP inhibitors failed in clinical trials [91] |
Case Study 1: Inflammatory Bowel Disease (IBD)
The IBD Therapeutic Challenge
Inflammatory bowel disease, encompassing Crohn's disease (CD) and ulcerative colitis (UC), illustrates the profound challenges in translational medicine. Despite the availability of new therapeutic modalities, complete remission is typically achieved and maintained in only approximately 30% of patients [92]. This limited success rate stems from the complex etiology of IBD, which involves multiple genetic loci combined with differential environmental exposures, suggesting that IBD represents a continuum of disorders rather than distinct homogeneous disease entities [92].
The current therapeutic approach to IBD relies predominantly on immunosuppression, which fails to account for basic disease variability. Treatments are prescribed based on statistical considerations related to the response of the average patient in clinical trials rather than on personal considerations [92]. This "one-size-fits-all" approach results in disappointing outcomes across different drug classes, including anti-TNF antibodies, anti-migration agents, and small molecules.
Success Stories in IBD Translation
Multi-Centric Biobanking Initiatives
One of the most successful translational strategies in IBD has been the establishment of structured, multi-centric biobanking initiatives. The Belgian IBD biobank network, encompassing university hospitals in Leuven, Ghent, Brussels, and Liège, has demonstrated how systematic collection of human biological materials can accelerate discovery and validation [93]. The Leuven biobank alone contains DNA and serum from >4,000 IBD patients, >3,000 unaffected relatives, and 1,300 healthy controls, creating a powerful resource for genetic and translational studies [93].
This coordinated approach has yielded substantial scientific output, with numerous publications in high-impact journals (Lancet, Nature, Nature Genetics) and has facilitated participation in international consortia that have identified over 200 genetic loci associated with IBD risk [93]. The correlation between biobank activity and scientific publications demonstrates the value of well-structured, prospective collection of human biological materials for translational success.
Biomarker-Driven Treatment Selection
Alternative approaches for therapy selection in IBD have shown promise by focusing on matching known physiologic defects with appropriate drug effects. For instance, genetic studies have demonstrated linkages between polymorphisms in autophagy-associated genes and Crohn's disease [92]. This knowledge creates opportunities for targeted therapeutic interventions, such as the use of rapamycin (which inhibits mTOR and enhances autophagy) in patients with specific genetic profiles affecting autophagy pathways [92].
Similarly, thiopurines have been shown to enhance innate immune function by inhibiting p21Rac1, leading to improved bacterial killing—an effect that may correct the immune deficiency state associated with the most prevalent genetic polymorphism in NOD2 in the Caucasian population [92]. These examples illustrate how understanding conserved biological processes can inform mechanism-based treatment selection.
Experimental Protocols: IBD Biobanking and Biomarker Validation
Multi-Centric Biobank Establishment
The successful Belgian IBD biobank network employed the following protocol:
- Standardized Collection Protocols: Implementation of uniform procedures for collection of serum, DNA, tissue biopsies, and fecal samples across participating centers [93].
- Clinical Data Annotation: Comprehensive clinical characterization including disease phenotype, treatment history, and outcomes using standardized classification systems [93].
- Quality Management: Operation according to a quality management system based on ISO 9001, complemented with biobank-specific ISO 20387 standards and ISBER Best Practices [93].
- Informed Consent Framework: Development of ethical frameworks ensuring appropriate patient consent for future research use [93].
- Centralized ICT Infrastructure: Implementation of a central ICT backbone enabling data integration and sharing across institutions while maintaining security and privacy [93].
Vedolizumab Response Prediction Study
A recent study investigating predictors of vedolizumab treatment success employed the following methodology:
- Patient Cohort: 181 IBD patients (106 UC, 75 CD) receiving at least three applications of vedolizumab at a German tertiary referral center [94].
- Data Collection: Electronic patient records were used to collect baseline parameters including clinical disease activity scores, disease extent, previous anti-TNF exposure, concurrent medications, and laboratory values [94].
- Endpoint Definition: Primary endpoint was clinical remission at week 17 (fifth vedolizumab application), defined as partial Mayo score ≤1 for UC or Harvey-Bradshaw Index ≤4 for CD [94].
- Statistical Analysis: Univariable analyses followed by multivariable logistic regression prediction models, with receiver-operator characteristic (ROC) analysis to assess model performance [94].
The study found that 36% of UC patients and 35% of CD patients achieved clinical remission at 17 weeks, with lower clinical disease activity at baseline predicting higher likelihood of success [94].
Case Study 2: Cancer Models
The Cancer Translation Problem
The failure rate in oncology drug development is particularly striking. Promising results from preclinical models rarely translate into clinical success, with 85% of early clinical trials for novel drugs failing, and cancer drugs representing the largest proportion of these failures [91]. Furthermore, fewer than one in five cancer clinical trials find their way to the peer-reviewed literature, generally due to negative findings [91].
This translational crisis stems from fundamental limitations in existing cancer models. Mouse models, the most commonly used in vivo system, are actually poor models for the majority of human diseases due to crucial genetic, molecular, immunologic, and cellular differences [91]. Among 4,000+ genes compared between humans and mice, researchers found that transcription factor binding sites differed between the species in 41% to 89% of cases [91].
Success Strategies in Oncology Translation
Advanced Model Systems
The development of more physiologically relevant cancer models represents a promising approach to improving translational success:
Patient-Derived Xenografts (PDX): These models, derived from freshly resected human tumor tissue implanted into immunodeficient mice, better recapitulate the characteristics of human cancer, including tumor progression and evolution [95] [96]. PDX models have played key roles in validating biomarkers including HER2, BRAF, and KRAS mutations [95].
Tumor Organoids and 3D Cultures: These 3D structures recapitulate the identity of the organ or tissue being modeled, retaining characteristic biomarker expression more effectively than two-dimensional cultures [95] [96]. Organoids have been used to effectively predict therapeutic responses and guide personalized treatment selection [95].
Ex Vivo Organotypic Cultures (EVOCs): These cultures retain the native microenvironment and architecture of the originating tumor, providing a powerful platform for drug testing [96]. A key advantage is the retention of the native tumor microenvironment, though they cannot be propagated long-term [96].
Integrated Multi-Omics Approaches
Rather than focusing on single targets, multi-omic approaches utilize multiple technologies (genomics, transcriptomics, proteomics) to identify context-specific, clinically actionable biomarkers that might be missed with single-platform approaches [95]. The depth of information obtained through these integrated strategies enables identification of biomarkers for early detection, prognosis, and treatment response, ultimately contributing to more effective clinical decision-making [95].
Experimental Protocols: Advanced Cancer Model Development
PDX and Organoid Biobank Establishment
A comprehensive study describing the development of a breast cancer PDX and organoid biobank employed this protocol:
- Tumor Collection: Fresh tumor tissue collected from patients with representative breast cancer subtypes, including endocrine-resistant, HER2+, treatment-refractory, and metastatic tumors [96].
- PDX Generation: Implantation of tumor fragments into immunodeficient mice, with sequential passaging to expand models [96].
- Organoid Derivation: Generation of PDX-derived organoid (PDxO) lines from established PDX models [96].
- Multi-Level Characterization: Comprehensive genomic, transcriptomic, and phenotypic analysis to validate that models retain original tumor characteristics [96].
- Drug Response Validation: In vitro drug screening using organoids with parallel in vivo validation in PDX models to confirm response concordance [96].
Functional Biomarker Validation
Strategies for robust biomarker validation include:
- Longitudinal Sampling: Repeated biomarker measurements over time to capture dynamic changes in response to treatment or disease progression, providing a more complete picture than single time-point measurements [95].
- Functional Assays: Moving beyond correlative biomarker analysis to demonstrate biological relevance through functional tests that assess biomarker activity in physiologically relevant contexts [95].
- Cross-Species Transcriptomic Analysis: Integration of data from multiple species and models to provide a comprehensive picture of biomarker behavior, helping to bridge the gap between preclinical models and human biology [95].
The Scientist's Toolkit: Essential Research Reagent Solutions
Table 3: Key Research Reagents and Platforms for Translational Studies
| Reagent/Platform | Function | Translational Application |
|---|---|---|
| Patient-Derived Xenografts (PDX) | In vivo models from fresh human tumors in immunodeficient mice | More accurate therapeutic response prediction; biomarker validation [95] [96] |
| Patient-Derived Organoids (PDO) | 3D in vitro structures from patient tumors retaining original characteristics | High-throughput drug screening; personalized treatment prediction [95] [96] |
| Ex Vivo Organotypic Cultures (EVOC) | Short-term culture of fresh tumor slices retaining native microenvironment | Functional drug response testing with preserved tumor-stroma interactions [96] |
| Multi-Omics Profiling Platforms | Integrated genomic, transcriptomic, proteomic analysis | Comprehensive biomarker identification; molecular subtyping [95] |
| CRISPR Screening Platforms | High-throughput gene editing for functional genomics | Target validation; synthetic lethality discovery; resistance mechanism elucidation |
| Lentiviral Barcoding Systems | Cellular barcoding for lineage tracing and clonal dynamics | Tracking tumor evolution; understanding therapy resistance [95] |
Integrated Analysis: Convergent Strategies for Translational Success
Cross-Disease Principles for Improved Translation
Despite the distinct pathophysiology of IBD and cancer, convergent strategies emerge for enhancing translational success:
Human-Derived Model Systems: Both fields benefit from moving away from traditional animal models toward human-derived systems that better recapitulate human disease biology. In IBD, multi-centric biobanking provides well-characterized human biological materials [93], while in oncology, PDX models and organoids offer more physiologically relevant platforms for therapeutic testing [95] [96].
Longitudinal Functional Validation: Static biomarker assessment is insufficient for robust translation. Both fields require longitudinal monitoring and functional validation to establish clinical utility [95]. In IBD, this means tracking biomarker dynamics throughout treatment courses [94], while in cancer, it involves assessing functional responses in complex model systems [96].
Multi-Omics Integration: The complexity of both IBD and cancer necessitates integrated analytical approaches that capture multiple dimensions of biological regulation. Genomic, transcriptomic, and proteomic datasets provide complementary insights that enhance predictive accuracy [95].
The Deep Homology Perspective: Implications for Target Selection
The deep homology framework suggests that targeting evolutionarily conserved pathways may offer more predictable translational outcomes, as these represent fundamental biological processes with minimal redundancy. However, this approach requires careful consideration of therapeutic index, as conserved pathways often play critical roles in normal physiology [8] [21]. The failures of MMP inhibitors and TGN1412 illustrate the risks associated with targeting conserved pathways without sufficient understanding of their pleiotropic functions [91].
Visualizing the Translational Workflow: Pathway Diagrams
Deep Homology in Translational Research
Integrated Translational Workflow
The case studies in inflammatory bowel disease and cancer models demonstrate both the profound challenges and promising strategies in clinical translation. The integration of human-relevant model systems, comprehensive molecular profiling, and longitudinal functional validation represents a convergent approach across therapeutic areas. Furthermore, the conceptual framework of deep homology provides valuable insights for target selection, emphasizing the potential advantages of targeting evolutionarily conserved biological pathways.
Moving forward, successful translation will require continued refinement of model systems to better capture human disease complexity, along with development of analytical frameworks that integrate multiple data dimensions to generate more accurate predictions of clinical utility. The establishment of collaborative networks and standardized biobanking resources, as demonstrated by the successful IBD initiatives, provides a template for accelerating translation across disease areas. Through these integrated approaches, the promise of precision medicine—delivering the right treatment to the right patient at the right time—may finally be realized.
The concept of deep homology describes the preservation of biological modules across vast evolutionary distances, where organisms share common genetic or protein machinery despite extensive sequence and structural divergence. In protein families, this phenomenon manifests as the conservation of molecular function amid substantial variation in amino acid sequences and three-dimensional structures. This paradox presents a fundamental challenge in evolutionary biology and drug development: how can core function persist while its molecular implementation changes so dramatically? Understanding these patterns is crucial for reliable protein function annotation, accurate reconstruction of evolutionary histories, and informed drug discovery efforts that target conserved functional sites.
Recent advances in structural biology, sequencing technologies, and computational methods have revealed that functional conservation often extends far beyond detectable sequence similarity, with profound implications for assessing deep homology. This technical guide examines the mechanisms, detection methods, and evolutionary patterns underlying this phenomenon, providing researchers with frameworks for investigating deep homology in protein families relevant to animal design and therapeutic development.
Quantitative Patterns of Long-Term Protein Evolution
Temporal Divergence Models and Limits
Comprehensive analysis of orthologous enzymes with identical molecular functions reveals that their sequence divergence follows predictable patterns over evolutionary timescales. When comparing orthologs across billions of years of evolution, the decline in sequence similarity significantly slows after approximately 1-2 billion years of independent evolution, eventually reaching an effective divergence limit above 25% sequence identity [97]. This stabilization occurs not because substitution rates decrease, but because only a limited number of amino acid types (typically 2-4 on average per site) can maintain the protein's molecular function, with back substitutions becoming increasingly common at extremely long timescales [97].
Three primary models describe these long-term divergence patterns. The first model represents unlimited divergence through exponential decay (y = 100 × e^(-R₀×t)), while the second incorporates a divergence limit (y = Y₀ + (100 - Y₀) × e^(-R₀×t)), where Y₀ represents the long-term sequence identity plateau. A third, more biologically realistic model incorporates variable substitution rates across sites using a gamma distribution [97]. For 62 of 64 enzymatic activities studied, the limited divergence model provided significantly better fit to empirical data than the unlimited divergence model, demonstrating that functional conservation imposes substantial constraints on long-term sequence evolution [97].
Table 1: Evolutionary Divergence Models for Protein Sequences
| Model Type | Mathematical Formula | Key Parameters | Biological Interpretation |
|---|---|---|---|
| Unlimited Divergence | y = 100 × e^(-R₀×t) | R₀: Initial substitution rate | Independent substitutions at all sites without functional constraints |
| Limited Divergence | y = Y₀ + (100 - Y₀) × e^(-R₀×t) | Y₀: Long-term identity limit, R₀: Initial substitution rate | Functional constraints limit divergence through limited acceptable amino acids per site |
| Variable Rate (Gamma Distributed) | y = 100 × (R₀×t/α + 1)^(-α) | α: Shape parameter of gamma distribution | Different protein sites evolve at different rates according to functional importance |
Practical Thresholds for Function Transfer
For researchers engaged in protein annotation and function prediction, practical sequence identity thresholds provide guidelines for reliable function transfer. Domain-based and family-specific thresholds significantly increase annotation reliability compared to generic whole-protein thresholds [98].
Table 2: Sequence Identity Thresholds for Reliable Protein Function Transfer
| Function Level | Whole-Protein Threshold | Domain-Based Threshold | Annotation Reliability |
|---|---|---|---|
| Third-level EC digits | 40% | 50% | >90% |
| Full EC number | 60% | 70% | >90% |
| KEGG/MIPS Functional Catalogue | - | 80% | High confidence |
These thresholds are particularly important for drug development targeting specific protein functions, as they help distinguish between conserved functional sites and structurally tolerated variation. Domain-level analysis provides more reliable function prediction because proteins often consist of multiple domains with different evolutionary histories and functional constraints [98].
Biophysical and Structural Mechanisms
Local Energetic Frustration Conservation
The concept of local energetic frustration offers a biophysical framework for understanding functional conservation despite sequence variation. According to the "Minimal Frustration Principle," naturally folding proteins have evolved to minimize strong energetic conflicts between residues in their native states, unlike random heteropolymers [99]. However, some localized conflicts are often preserved due to functional requirements, creating a trade-off between molecular function and local stability.
FrustraEvo, a methodology for analyzing local frustration conservation within protein families, quantifies how well optimized for folding a given residue-residue interaction is compared to random interactions in non-native conformations [99]. This approach measures the conservation of local frustration patterns across multiple sequence alignments, identifying residues under strong evolutionary constraint. Highly frustrated residues often cluster around functional sites like ligand-binding pockets, catalytic centers, and protein-protein interaction interfaces, while minimally frustrated residues typically form the structurally stable protein core [99].
In studies comparing frustration conservation (FrustIC) with sequence conservation (SeqIC), frustration conservation showed stronger correlation with experimental measurements of protein stability and function from deep mutational scanning experiments. For SH3 and PDZ domains, FrustIC correlated with stability measurements at r = -0.79 to -0.82, compared to r = -0.63 to -0.69 for SeqIC [99]. This demonstrates that frustration conservation provides a more accurate predictor of functional constraints than sequence conservation alone.
Structural Determinants Amid Divergence
Structural analysis of deeply homologous proteins reveals several patterns that enable functional conservation amid divergence. First, the overall architectural fold often shows greater conservation than sequence, with the spatial arrangement of key functional residues maintained despite extensive sequence variation [97]. Second, core structural elements surrounding active sites typically show higher conservation than peripheral regions, creating a structurally conserved functional "scaffold" [99]. Third, even when global structures diverge, local microenvironments around functional sites often retain similar physicochemical properties through different amino acid combinations [97].
These principles explain how proteins can maintain identical molecular functions (such as catalyzing the same biochemical reaction) while their sequences and structures diverge substantially. The conservation of key functional residues, maintenance of overall structural topology around active sites, and preservation of local physicochemical microenvironments collectively enable functional conservation across evolutionary timescales [97].
Methodological Framework for Analysis
Experimental and Computational Workflows
Investigating deep homology requires integrated methodologies combining evolutionary analysis, structural comparison, and functional validation. The following workflow outlines key experimental and computational approaches for assessing functional conservation and structural divergence in protein families.
Diagram 1: Deep Homology Assessment Workflow
Research Reagent Solutions
Table 3: Essential Research Reagents and Resources for Deep Homology Studies
| Reagent/Resource | Function in Analysis | Application Examples |
|---|---|---|
| CATH-Gene3D | Protein domain family classification | Identifying homologous domains, functional annotation transfer [98] |
| FrustraEvo Algorithm | Local frustration conservation analysis | Identifying biophysically constrained residues in protein families [99] |
| ddPCA (Double-deep Protein Fragment Complementation Assay) | High-throughput measurement of variant effects on stability and function | Quantifying sequence-structure-function relationships in protein families [99] |
| eggNOG Orthologous Groups | Functional annotation of evolutionary relationships | Tracing functional conservation across taxonomic groups [100] |
| Synteny-Based Algorithms (e.g., IPP) | Identifying orthologous regulatory regions beyond sequence similarity | Detecting conserved non-coding elements with divergent sequences [101] |
| Sequence Similarity Networks | Visualizing and analyzing remote homology relationships | Identifying divergent protein variants in metagenomic data [100] |
Case Studies in Protein Families
Enzyme Orthologs Across Deep Time
Analysis of 64 enzyme activities across 22 species spanning the three domains of life provides compelling evidence for deep homology maintained under functional constraints. After 3-4 billion years of evolution, orthologous enzymes with identical molecular functions maintain sequence identities substantially above random expectation (>25% vs. ~13.5% for random sequences) [97]. This conservation is not primarily driven by universally conserved sites but emerges from the limited repertoire of acceptable amino acids at each position, with only 2-4 amino acid types accepted on average per site across orthologous sequences [97].
The divergence patterns also differ significantly between protein sites with small versus large fitness effects. Sites with large fitness effects show stronger conservation and less divergence over time, while sites with small fitness effects accumulate more substitutions, contributing to structural divergence while maintaining core function [97]. This demonstrates how functional conservation operates through a limited set of biophysical constraints rather than requiring extensive sequence similarity.
RAS Superfamily Divergence Patterns
The RAS superfamily exemplifies how local frustration conservation identifies functional constraints despite sequence divergence. Analysis of KRAS frustration patterns revealed that frustration conservation (FrustIC) correlated better with experimental stability measurements (r = -0.47) than sequence conservation (SeqIC), which showed no significant correlation [99]. This demonstrates that biophysical constraints preserve functional sites even when sequence similarity becomes minimal.
Notably, KRAS contains one highly frustrated conserved position (K117) that interacts with nucleotide substrates, illustrating how functional requirements can maintain locally unfavorable energetic configurations across evolutionary timescales [99]. This preservation of highly frustrated interactions suggests strong positive selection for functional reasons, creating a trade-off between stability and molecular function that is characteristic of deep homology.
Metagenomic Discovery of Divergent Variants
Iterative network-based approaches have revealed highly divergent proteins within ancient, conserved gene families from environmental metagenomes. Using sequence similarity networks to probe 40 million oceanic ORFs, researchers identified environmental variants of 53 core gene families with divergence comparable to the differences between Archaea and Bacteria [100]. These included novel structural variants of essential SMC (Structural Maintenance of Chromosomes) genes, divergent polymerase subunits forming deep-branching clades, and variant DNA recombinases in Bacteria and viruses [100].
This "microbial dark matter" represents significant uncharacterized diversity within well-studied protein families, suggesting that current knowledge of protein sequence space remains substantially incomplete. The discovery of these highly divergent yet functional variants through iterative homology detection highlights the potential for identifying new deep homology relationships beyond conventional sequence-based methods [100].
Implications for Drug Development
Targeting Conserved Functional Sites
Understanding deep homology patterns provides powerful strategies for drug development, particularly for targeting conserved functional sites across protein families. The limited number of acceptable amino acids per site in functionally conserved regions [97] suggests that these sites present attractive targets with reduced potential for drug resistance through mutation. Additionally, the conservation of local frustration patterns around functional sites [99] indicates that these regions maintain specific physicochemical properties ideal for small molecule interactions.
Drug development pipelines can leverage deep homology principles to identify functionally constrained sites that are conserved across pathogen variants or related host protein families. This approach enables designing broad-spectrum therapeutics less susceptible to resistance mutations while minimizing off-target effects through precise targeting of functional microenvironments preserved across evolutionary timescales.
Predicting Functional Transfer in Novel Targets
For drug developers working with newly identified targets, deep homology assessment provides frameworks for predicting functional conservation in the absence of extensive experimental data. Domain-based sequence identity thresholds [98] offer practical guidelines for inferring molecular function, while frustration conservation analysis [99] helps identify functionally critical residues that may not exhibit high sequence conservation.
These approaches are particularly valuable for prioritizing targets from metagenomic discoveries [100] or poorly characterized protein families. By identifying deeply homologous relationships to well-studied protein families, researchers can leverage existing knowledge about mechanism of action, potential inhibitors, and functional residues to accelerate target validation and drug discovery efforts.
The integration of homology modeling and the principles of deep homology represents a transformative approach in early drug discovery. This whitepaper provides an in-depth technical guide on leveraging computational protein models to assess the druggability of biological targets and predict their response in animal models. By framing these methodologies within the context of deep homology, we elucidate how conserved genetic regulatory mechanisms across distantly related species can inform the validity of animal models for human disease. The document offers a detailed roadmap for researchers, featuring structured quantitative data, explicit experimental protocols, and specialized visualization tools to enhance the predictive accuracy of target suitability assessments, thereby de-risking the drug development pipeline.
The pursuit of novel therapeutics is a protracted and costly endeavor, often requiring over a decade and exceeding $2 billion in investment to bring a single drug to market [102]. A significant point of attrition is the failure of targets identified in early research to translate successfully to clinical efficacy, often due to inadequate validation and a poor understanding of their behavior across different biological systems [103] [104]. Within this challenging landscape, target identification and validation constitute the critical foundation, confirming a target's direct role in a disease pathway and its potential for therapeutic modulation [104]. A crucial, subsequent question is that of druggability—whether the target's structure possesses a binding site that can be effectively and safely targeted by a small molecule or biologic.
Homology modeling, a computational technique for predicting a protein's three-dimensional structure from its amino acid sequence based on similarity to experimentally solved templates, has emerged as a powerful tool for early druggability assessment [30]. Its utility is grounded in the observation that protein structure is evolutionarily more conserved than amino acid sequence. Concurrently, the concept of deep homology provides a framework for understanding the conservation of genetic regulatory mechanisms—the "toolkits" for building anatomical structures—across vastly different species [8]. This principle explains why distantly related organisms, such as arthropods and chordates, can use remarkably similar gene regulatory networks (kernels or Character Identity Networks - ChINs) to pattern non-homologous organs, like their circulatory systems [8] [83]. For drug discovery, this implies that the biological role and interaction potential of a protein target may be conserved in animal models, even if the resulting anatomical structures are not directly comparable. This whitepaper synthesizes these concepts into a cohesive technical guide, providing researchers with the methodologies to build and utilize homology models for predicting target suitability and cross-species response.
Theoretical Foundation: Deep Homology and Target Conservation
The traditional definition of homology refers to a shared ancestry between morphological structures. However, the advent of evolutionary developmental biology (evo-devo) revealed that even morphologically distinct structures can be governed by deeply conserved genetic circuits. This phenomenon is termed deep homology [8] [83].
- Character Identity Networks (ChINs): A ChIN is a core set of gene regulatory interactions that defines a specific morphological character or module [8]. These networks are evolutionarily more stable than the individual genes themselves and are repeatedly re-deployed during embryogenesis. The presence of a shared ChIN underlying the development of an organ in two different species provides a mechanistic basis for historical homology, even if the final character states (e.g., the shape of a limb) differ.
- Kernels and Deep Evolutionary Conservation: At a deeper phylogenetic level, "kernels" are sub-circuits of gene regulatory networks that are central to body plan patterning and are highly refractory to evolutionary change. A canonical example is the conservation of the genetic toolkit for heart specification between arthropods and chordates, two phyla that diverged over 500 million years ago [8]. This indicates a common regulatory blueprint for a primitive circulatory organ at the base of the Bilateria.
- Implications for Drug Discovery: From a druggability perspective, deep homology suggests that if a target protein is a key component of a conserved ChIN or kernel, its functional role, its interactions with other biomolecules, and its structural binding sites are more likely to be conserved across a wide range of species, including standard animal models. This conservation increases confidence that in vitro and in vivo findings in these models will translate to human biology. Conversely, a target lacking such deep conservation may exhibit significant functional differences between species, rendering animal model data less predictive [105].
Homology Modeling: A Technical Guide for Druggability Assessment
Homology modeling, or comparative modeling, is the most accurate method for predicting a protein's 3D structure when no experimental structure is available [30]. The process is multi-staged, and each step is critical for generating a reliable model for druggability assessment.
The Homology Modeling Workflow
The following diagram illustrates the standard, iterative workflow for developing a high-quality homology model.
Detailed Experimental Protocols
Protocol 1: Template Identification and Alignment
Objective: To identify a suitable experimental protein structure as a template and generate an optimal sequence alignment.
- Template Search: Submit the target amino acid sequence to search servers against the Protein Data Bank (PDB).
- Template Selection Criteria: Prioritize templates based on:
- Sequence Identity: A higher percentage generally yields a more accurate model (see Table 1).
- Coverage: The template should cover the entire target sequence, especially functional domains.
- Experimental Resolution: A higher-resolution X-ray crystal structure (e.g., <2.0 Å) is preferable.
- Ligand Presence: Templates co-crystallized with a relevant ligand are invaluable for binding site analysis.
- Sequence Alignment: Perform a multiple sequence alignment using tools like ClustalW, T-Coffee, or PROBCONS to optimize the target-template alignment, which is the single most critical factor for model accuracy [30].
Protocol 2: Model Building, Refinement, and Validation
Objective: To construct an all-atom model and rigorously evaluate its quality.
- Model Building: Utilize automated software for initial model construction. Common methods include:
- Spatial Restraint: As implemented in MODELER, which satisfies spatial restraints derived from the template [30].
- Segment Matching: Assembling the model from short fragments of known structures that fit the target sequence.
- Loop and Side-Chain Modeling: Regions with insertions/deletions (loops) and side chains are often modeled separately using conformational sampling and rotamer libraries.
- Energy Minimization: Subject the initial model to energy minimization using molecular mechanics force fields (e.g., CHARMM, AMBER) to relieve steric clashes and geometric strain [30].
- Model Validation: Critically assess the model's structural quality using:
- Stereo-chemical Checks: PROCHECK/MolProbity for Ramachandran plot analysis, bond lengths, and angles.
- Statistical Potential Scores: Verify3D and ERRAT to evaluate the compatibility of the 3D model with its own amino acid sequence.
- MD Simulations: Short molecular dynamics simulations in explicit solvent can assess model stability under near-physiological conditions [30].
Model Quality and Druggability Metrics
The utility of a homology model in drug discovery is directly correlated with its quality, which is itself a function of target-template sequence identity. The table below summarizes key benchmarks and applications.
Table 1: Homology Model Quality and Application Guide
| Sequence Identity to Template | Expected Model Accuracy | Recommended Applications in Drug Discovery |
|---|---|---|
| >50% | High | Virtual screening, De novo ligand design, Detailed binding site analysis [30] |
| 25% - 50% | Medium | Guiding mutagenesis experiments, Qualitative binding site mapping, Low-throughput in silico screening [30] |
| <25% | Low (Tentative) | Low-resolution hypothesis generation; requires strong experimental validation [30] |
Predicting Animal Model Response via Deep Homology Analysis
Once a reliable homology model is built, its value is extended by integrating it with deep homology principles to gauge the likely translational relevance of animal models.
Integrated Workflow for Cross-Species Prediction
The following diagram outlines a logical workflow for integrating homology modeling with deep homology analysis to assess target suitability and animal model predictability.
Experimental Protocol for Cross-Species Assessment
Objective: To evaluate the structural and functional conservation of a drug target across species to validate the choice of animal model.
- Ortholog Identification: Use genomic databases (e.g., Ensembl, NCBI) to identify the primary orthologs of the human target in candidate animal models (e.g., mouse, rat, zebrafish).
- Comparative Modeling: Build homology models for the key orthologs using the same protocol applied to the human target.
- Binding Site Comparison: Structurally align the human and ortholog models. Quantitatively compare the binding sites using metrics like:
- Volume and Shape Overlap: Using tools like CASTp or POCASA.
- Residue Conservation: Analyzing the conservation of key amino acids known to be critical for ligand binding or function.
- Deep Homology Interrogation:
- Literature/Data Mining: Search for evidence that the target gene is part of a conserved ChIN or kernel (e.g., through gene expression databases, RNA-seq data from model organisms).
- Functional Assays: If the target is a member of a deeply conserved network (e.g., a key signaling pathway like Hedgehog or Wnt), confidence in the animal model's predictive power increases significantly [8].
- Integrated Prediction:
- High-Fidelity Model: An animal model where the ortholog has a highly conserved binding site and the target is part of a deeply homologous network is predicted to have high translational fidelity.
- Low-Fidelity Model: Significant structural divergence in the binding site or absence of deep homology indicates that results from that animal model may not reliably predict human response, necessitating caution and the use of human-relevant models like organ-on-a-chip or organoids [105].
The Scientist's Toolkit: Essential Research Reagents and Solutions
Successful implementation of the described protocols relies on a suite of specialized computational and biological tools.
Table 2: Key Research Reagent Solutions for Integrated Assessment
| Category | Tool/Reagent | Primary Function |
|---|---|---|
| Template Identification | BLAST, PSI-BLAST, HMMER | Identify homologous protein structures from the PDB for use as modeling templates [30]. |
| Sequence Alignment | ClustalW, T-Coffee, MUSCLE | Generate accurate multiple sequence alignments between target and template sequences [30]. |
| Model Building | MODELLER, SWISS-MODEL, I-TASSER | Build initial 3D protein models based on template structures and sequence alignments [30]. |
| Model Validation | PROCHECK, MolProbity, Verify3D | Assess the stereochemical quality and structural rationality of the generated homology models [30]. |
| Deep Homology Analysis | RNA Interference (RNAi), CRISPR-Cas9 | Validate target function and its role in conserved gene networks in model organisms [103] [104]. |
| Human-Relevant Validation | Organ-on-a-Chip, Human Organoids | Test compound efficacy and toxicity in vitro using human cells, bypassing species-specific limitations [105]. |
The strategic confluence of homology modeling and deep homology principles provides a powerful, rational framework for de-risking the early stages of drug discovery. By generating reliable structural models of therapeutic targets, researchers can computationally assess their druggability before committing extensive resources. Furthermore, by evaluating these targets through the lens of deep homology—assessing their membership in evolutionarily conserved genetic circuits—scientists can make more informed predictions about the translational fidelity of animal models. This integrated approach, leveraging in silico tools, functional genomics, and emerging human-relevant assays like organs-on-chips, promises to enhance the efficiency of the drug development pipeline, reducing the high failure rates associated with poor target selection and inadequate model systems.
The concept of deep homology—the sharing of ancient genetic regulatory apparatus across distantly related species—has revolutionized evolutionary developmental biology and disease mechanism research. This principle posits that despite dramatic morphological diversification, fundamental genetic circuits and cell types are conserved across vertebrate evolution and can be leveraged to understand human disease pathogenesis. Cross-species validation provides a powerful methodological framework for testing functional conservation of these genetic networks, from fundamental developmental processes to complex disease mechanisms. This technical guide examines current methodologies, experimental protocols, and analytical frameworks for implementing robust cross-species validation in biomedical research, with particular emphasis on applications in neuroscience, respiratory medicine, and neurodegenerative disorders.
The theoretical foundation of this approach stems from findings that despite 450 million years of evolutionary divergence, core genetic programs maintain remarkably similar functions. Recent studies on the teleost telencephalon demonstrate striking transcriptional similarities between cell-types in the fish telencephalon and subpallial, hippocampal, and cortical cell-types in tetrapods, supporting partial eversion of the teleost telencephalon [4]. Similarly, research on FoxP2 reveals this transcription factor and its regulatory network shapes neural plasticity in cortico-basal ganglia circuits underlying sensory-guided motor learning across diverse taxa, illustrating how complex cognitive traits 'descend with modification' [3]. These conserved molecular pathways operating across evolutionary timescales provide the fundamental rationale for cross-species validation approaches in modern biomedical research.
Core Methodological Frameworks for Cross-Species Validation
Integrated Transcriptomic Analysis
Contemporary cross-species validation employs sophisticated transcriptomic integration to identify conserved genetic signatures. This typically involves RNA sequencing of model systems combined with computational analysis of publicly available datasets from both model organisms and human clinical samples. A representative workflow from acute respiratory distress syndrome (ARDS) research illustrates this approach: researchers conducted RNA sequencing on LPS-stimulated MH-S cells (a mouse alveolar macrophage cell line) and integrated these data with publicly available murine (GSE225406) and human (GSE40885) alveolar macrophage datasets to identify conserved differentially expressed genes (DEGs) across species [106]. This integrated analysis revealed 45 conserved upregulated genes and 4 downregulated genes across species, highlighting core transcriptional regulators of LPS-induced macrophage activation.
The power of transcriptomic integration lies in its ability to distinguish species-specific responses from evolutionarily conserved mechanisms. In the ARDS study, functional enrichment analysis of conserved DEGs revealed significant involvement of immune-inflammatory pathways, while protein-protein interaction network analysis identified 10 core genes potentially central to pathogenesis [106]. This systematic approach to transcriptomic conservation provides a template for identifying robust, evolutionarily conserved disease mechanisms rather than species-specific artifacts.
Cross-Species miRNA Profiling and Validation
MicroRNAs have emerged as promising candidates for neurological biomarkers due to their stability in circulation, tissue-specific expression patterns, and roles in regulating key cellular processes implicated in neurodegeneration. Cross-species miRNA validation follows a systematic workflow beginning with temporal profiling in animal models, proceeding through robust feature selection, and culminating in validation across multiple human cohorts.
A representative Parkinson's disease study implemented this approach through temporal miRNA profiling in an acute MPTP mouse model, comparing day 0 versus day 5 post-treatment [107]. To address high-dimensional small-sample challenges, researchers employed global permutation testing and stability selection with elastic net regularization over 2,000 iterations. This analytical rigor yielded a compact 6-miRNA panel comprising miR-92b, miR-133a, miR-326, miR-125b, miR-148a, and miR-30b that was subsequently validated in three independent human cohorts representing different sample types [107]. The demonstrated platform-agnostic stability of this signature, performing comparably in PBMC and serum exosomes despite biological and technical differences, highlights the power of systematic cross-species miRNA validation.
Mendelian Randomization and Genetic Correlation Analysis
Mendelian randomization (MR) has emerged as a powerful method for validating putative therapeutic targets identified in model organisms. This approach uses genetic variants as instrumental variables to test causal relationships between exposures and outcomes, providing human genetic evidence to complement experimental findings from model systems.
In a study investigating amlodipine as a novel ADHD treatment, researchers performed cross-species analysis in SHR rats and adgrl3.1-/- zebrafish demonstrating the drug's efficacy in reducing hyperactivity and impulsivity [108]. Crucially, they then employed Mendelian randomization analysis linking ADHD to genetic variations in L-type calcium channel subunits (α1-C; CACNA1C, β1; CACNB1, α2δ3; CACNA2D3) targeted by amlodipine [108]. This combination of cross-species behavioral pharmacology with human genetic validation provides a compelling framework for translational research, effectively bridging the gap between animal models and human disease mechanisms.
Table 1: Cross-Species Validation Methodologies and Applications
| Methodology | Key Features | Representative Application | Advantages |
|---|---|---|---|
| Integrated Transcriptomics | RNA-seq across species, DEG identification, functional enrichment | ARDS alveolar macrophage conserved genes [106] | Distinguishes conserved from species-specific mechanisms |
| miRNA Profiling | Temporal expression analysis, stability selection, multi-cohort validation | Parkinson's disease 6-miRNA blood signature [107] | Platform-agnostic biomarkers; high stability |
| Mendelian Randomization | Genetic instrumental variables, polygenic risk scores, causal inference | ADHD and L-type calcium channels [108] | Provides human genetic evidence for mechanistic links |
| Cross-Species Behavioral Pharmacology | Multiple model organisms, validated behavioral paradigms | Amlodipine in ADHD rat and zebrafish models [108] | Tests functional conservation of therapeutic mechanisms |
Experimental Protocols for Cross-Species Validation
Protocol 1: Cross-Species Transcriptomic Conservation Analysis
Objective: Identify conserved gene expression signatures across species for a disease of interest.
Sample Collection and Preparation:
- Collect target tissue/cells from at least two species (typically mouse and human) under comparable experimental conditions
- For the ARDS study, researchers used: (1) LPS-stimulated MH-S mouse alveolar macrophage cell line; (2) Primary mouse alveolar macrophages from GEO dataset GSE225406; (3) Human alveolar macrophages from GEO dataset GSE40885 [106]
- Ensure standardized RNA extraction protocols across all samples (e.g., miRNeasy Serum/Plasma Kit for miRNA, Cell/Bacterial Total RNA Extraction Kit for mRNA)
- Assess RNA quality using Agilent Bioanalyzer with RNA Integrity Number (RIN) ≥ 7 as quality threshold
Library Preparation and Sequencing:
- Perform library construction using poly-A selection for mRNA or size selection for small RNAs
- Utilize Illumina platforms (e.g., HiSeq 2500) for high-throughput sequencing
- Sequence at sufficient depth (typically ≥20 million reads per sample for RNA-seq)
Computational Analysis:
- Quality control of raw sequencing data using FastQC
- Alignment to respective reference genomes (e.g., GRCh38 for human, GRCm39 for mouse)
- Differential expression analysis using limma R package with |Fold-change| > 1.5 and p < 0.05 thresholds [107] [109]
- Identification of conserved DEGs appearing in both species datasets
- Functional enrichment analysis using clusterProfiler for GO and KEGG pathways [109]
- Protein-protein interaction network construction using STRING database (interaction score > 0.400) [109]
Validation:
- Experimental validation using qRT-PCR, Western blot, immunohistochemical staining
- Assessment of diagnostic potential using clinical datasets (e.g., AUC calculation via ROC analysis)
Protocol 2: Cross-Species Behavioral Pharmacology with Genetic Validation
Objective: Validate therapeutic mechanisms across species and confirm with human genetic evidence.
Animal Models and Behavioral Testing:
- Utilize multiple model organisms with complementary advantages (e.g., zebrafish for high-throughput screening, rats for complex behavioral assessment)
- In the ADHD study, researchers used: (1) Spontaneously Hypertensive Rats (SHR) with WKY rats as controls; (2) adgrl3.1-/- zebrafish mutant model [108]
- Implement standardized behavioral tests: Open Field Test for general hyperactivity in rats; 5-Choice Serial Reaction Time Task for impulsivity in zebrafish
- Ensure blinded drug administration and behavioral scoring
- Maintain appropriate sample sizes (n=20 per group for rat studies) based on power analysis
Drug Treatment:
- Administer test compounds with appropriate vehicle controls
- For the ADHD study: 30-day administration of amlodipine (10 mg/kg) via intraperitoneal injection in rats; 30-minute immersion in 10 µM amlodipine for zebrafish [108]
- Include positive controls (e.g., methylphenidate for ADHD models)
Genetic Validation in Human Populations:
- Perform Mendelian randomization using large-scale GWAS summary statistics
- Test causal relationships between drug targets (e.g., L-type calcium channel genes) and disease risk
- Calculate polygenic risk scores to test for interaction between genetic liability and drug effects
- In the ADHD study, MR linked ADHD to variations in CACNA1C, CACNB1, and CACNA2D3 [108]
Protocol 3: Cross-Species miRNA Biomarker Development
Objective: Develop conserved miRNA signatures from animal models to human applications.
Animal Model Selection and Temporal Profiling:
- Select pathophysiologically relevant animal models (e.g., MPTP mouse model for Parkinson's disease)
- Implement temporal sampling strategy (e.g., day 0 baseline vs. day 5 post-treatment in MPTP model) [107]
- Collect appropriate biospecimens (serum, plasma, tissue) matching intended human application
miRNA Expression Profiling:
- Extract RNA using specialized kits for small RNAs (e.g., miRNeasy Serum/Plasma Kit)
- Perform miRNA expression profiling using appropriate platforms (e.g., Affymetrix GeneChip miRNA 4.0 array)
- Normalize data using robust multichip average or similar algorithms
Statistical Analysis and Feature Selection:
- Conduct differential expression analysis using limma with FDR correction (FDR <0.05) [107]
- Address high-dimensionality challenges through global permutation testing (e.g., 5,000 permutations)
- Implement stability selection with elastic net regularization over multiple iterations (e.g., 2,000 iterations) to derive compact biomarker panels
Cross-Species and Cross-Platform Validation:
- Validate animal-derived signatures in multiple independent human cohorts representing different sample types (e.g., PBMC, serum exosomes)
- Assess performance using ROC analysis with permutation-based p-values
- Require consistent discriminative performance across platforms (typical AUC >0.70 considered acceptable) [107]
Visualization of Cross-Species Validation Workflows
Integrated Transcriptomic Analysis Workflow
Integrated Transcriptomic Analysis for Cross-Species Validation
Cross-Species miRNA Biomarker Development
Cross-Species miRNA Biomarker Development Pipeline
Table 2: Essential Research Reagents for Cross-Species Validation Studies
| Reagent/Resource | Specifications | Application | Example Use |
|---|---|---|---|
| miRNA Profiling Arrays | Affymetrix GeneChip miRNA 4.0 (3,163 miRNA probes) | Comprehensive miRNA expression profiling | Temporal miRNA changes in MPTP mouse model [107] |
| RNA Extraction Kits | miRNeasy Serum/Plasma Kit (Qiagen) | High-quality small RNA isolation from biofluids | Serum miRNA analysis in Parkinson's study [107] |
| Animal Disease Models | MPTP mouse model (Parkinson's), SHR rat (ADHD), adgrl3.1-/- zebrafish | Pathophysiologically relevant systems | Cross-species therapeutic validation [107] [108] |
| Behavioral Testing Apparatus | Med Associates Open Field Test chambers, Zantiks AD 5-CSRTT system | Standardized behavioral phenotyping | Impulsivity measurement in zebrafish [108] |
| Bioinformatics Tools | limma R package, WGCNA, clusterProfiler | Differential expression and functional analysis | Conserved DEG identification [107] [109] [106] |
| Public Genomic Databases | GEO datasets (human and mouse), STRING database | Data integration and PPI network analysis | Cross-species transcriptomic integration [109] [106] |
| Validation Reagents | qPCR assays, Western blot antibodies, IHC reagents | Experimental confirmation of findings | Core gene validation in ARDS study [106] |
Quantitative Data Synthesis in Cross-Species Research
Table 3: Performance Metrics in Cross-Species Validation Studies
| Study Focus | Model Systems | Key Findings | Performance Metrics | Clinical Translation |
|---|---|---|---|---|
| Parkinson's Disease miRNA Signature | MPTP mouse → Human PBMC and serum exosomes | 6-miRNA panel derived from mouse model | Mouse: 17 significant miRNAs (FDR<0.05)Human: AUC = 0.791 (serum exosomes)AUC = 0.725 (serum exosomes 2020) [107] | Cross-platform stability supports clinical potential |
| ADHD Therapeutic Target Validation | SHR rats + adgrl3.1-/- zebrafish → Human genetics | Amlodipine efficacy across species + MR validation | Rat: reduced hyperactivity in OFTZebrafish: reduced hyperactivity/impulsivityHuman: MR links ADHD to CACNA1C, CACNB1, CACNA2D3 [108] | Genetic evidence supports target engagement in humans |
| ARDS Conserved Macrophage Genes | Mouse MH-S cells → Human alveolar macrophages | 45 conserved upregulated, 4 downregulated genes | Cross-species conserved DEGs identifiedDiagnostic potential: AUC = 0.86 in clinical datasets [106] | Core genes represent potential therapeutic targets |
| COPD-T2DM Shared Genetics | Human datasets → Mouse model validation | Shared DEGs (KIF1C, CSTA, GMNN, PHGDH) | Random forest/LASSO identified 6 critical genesXGBoost model: AUC = 0.996 for COPD prediction [109] | Machine learning enhances cross-species prediction |
Discussion and Future Directions
Cross-species validation represents a powerful paradigm for bridging the translational gap between basic research and clinical applications. By leveraging deep homology—the conservation of genetic circuits across evolution—researchers can distinguish fundamental disease mechanisms from species-specific epiphenomena. The methodologies outlined in this technical guide provide a systematic framework for implementing robust cross-species validation across diverse research contexts.
The future of cross-species validation will likely involve even more sophisticated integration of multi-omics data, single-cell technologies, and advanced machine learning approaches. As spatial transcriptomics becomes more accessible, researchers will be able to validate not just molecular signatures but conserved cellular ecosystems across species [4]. Similarly, emerging technologies for studying extracellular vesicles and cross-species RNA communication may reveal novel mechanisms of disease pathogenesis and potential therapeutic avenues [110].
Ultimately, the rigorous application of cross-species validation principles accelerates the identification of robust disease mechanisms and viable therapeutic targets. By demanding conservation across evolutionary distance, this approach provides a powerful filter for prioritizing the most promising candidates for further development, potentially reducing the high attrition rates that plague translational research. As the tools and datasets available for these analyses continue to expand, cross-species validation will remain an essential component of the biomedical research toolkit, firmly grounded in the principles of deep homology and evolutionary conservation.
Conclusion
Deep homology provides a powerful unifying framework that connects evolutionary biology with modern drug discovery. The conservation of genetic regulatory mechanisms across distantly related species, from signaling pathways to gene regulatory networks, offers unprecedented opportunities for understanding disease etiology and identifying therapeutic targets. Methodological advances in sequencing, protein structure prediction, and sensitive homolog detection are rapidly closing the sequence-structure gap, enabling more accurate exploitation of these evolutionarily conserved systems. However, successful translation requires careful attention to model selection, statistical validation, and recognition of both the powers and limitations of homology-based approaches. Future directions will likely involve the integration of deep homology concepts with multi-omics data, advanced AI-driven protein modeling, and the systematic application of quality assessment frameworks to improve preclinical-to-clinical translation. For drug development professionals, embracing this evolutionary perspective promises to enhance target prioritization, rational drug design, and ultimately, the success rate of therapeutic candidates.
References
- 1. Owen's Vertebral Archetype and Evolutionary Genetics-A ... [https://muse.jhu.edu/article/403553/summary]
- 2. Theories, laws, and models in evo‐devo - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC9292786/]
- 3. Evo-devo, deep homology and FoxP2: implications for the ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3130369/]
- 4. Spatially resolved cell atlas of the teleost telencephalon ... [https://www.nature.com/articles/s42003-024-06315-1]
- 5. Protein remote homology detection and structural ... [https://www.nature.com/articles/s41587-023-01917-2]
- 6. Fast, sensitive detection of protein homologs using deep ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12167706/]
- 7. Rprot-Vec: a deep learning approach for fast protein structure ... [https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-025-06213-1]
- 8. Deep homology in the age of next-generation sequencing - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC5182409/]
- 9. The developmental genetics of homology [https://pubmed.ncbi.nlm.nih.gov/17486120/]
- 10. Why call it developmental bias when it is just development? [https://biologydirect.biomedcentral.com/articles/10.1186/s13062-020-00289-w]
- 11. Developmental Constraints - Developmental Biology - NCBI [https://www.ncbi.nlm.nih.gov/books/NBK10032/]
- 12. Gene family innovation, conservation and loss on the ... [https://elifesciences.org/articles/34226]
- 13. The Notch signalling system: recent insights into the ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC4369923/]
- 14. The Notch pathway in Metazoa: a comparative analysis across ... [https://evodevojournal.biomedcentral.com/articles/10.1186/s13227-025-00246-6]
- 15. Notch signaling pathway: architecture, disease, and ... [https://www.nature.com/articles/s41392-022-00934-y]
- 16. Evolution of hedgehog and hedgehog-related genes, their ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC2362128/]
- 17. The evolutionary origin of hedgehog proteins [https://www.sciencedirect.com/science/article/pii/S0960982207017861]
- 18. The Hedgehog Signaling Pathway: Where Did It Come From? [https://journals.plos.org/plosbiology/article?id=10.1371/journal.pbio.1000146]
- 19. Evolutionary conservation of Wnt signaling [https://wnt.stanford.edu/evolutionary-conservation-wnt-signaling]
- 20. Deep homology and the origins of evolutionary novelty [https://pubmed.ncbi.nlm.nih.gov/19212399/]
- 21. Deep homology and design: a new series [https://sfmatheson.blogspot.com/2009/06/deep-homology-and-design-new-series.html]
- 22. a model for pattern formation within the vertebrate body plan [https://pubmed.ncbi.nlm.nih.gov/8763496/]
- 23. Development and the evolvability of human limbs - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC2840520/]
- 24. A gene regulatory network for specification and ... [https://www.sciencedirect.com/science/article/abs/pii/S0012160625000697]
- 25. How insights from cardiovascular developmental biology have ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3409324/]
- 26. Developmental and Evolutionary Heart Adaptations ... [https://www.mdpi.com/2308-3425/12/3/83]
- 27. The evolution of developmental biology through ... [https://www.sciencedirect.com/science/article/pii/S0092867424006329]
- 28. in Biology--A Problem for Naturalistic Science Homology [https://trueorigin.org/homology.php]
- 29. The application of irreversible genomic states to define and ... [https://evodevojournal.biomedcentral.com/articles/10.1186/s13227-025-00242-w]
- 30. Homology Modeling a Fast Tool for Drug Discovery [https://pmc.ncbi.nlm.nih.gov/articles/PMC3507339/]
- 31. Higher frequency of homologous chromosome pairing in ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11956967/]
- 32. 332. Examples of scientific data visualization with ... [https://medium.com/@ilakk2023/examples-of-scientific-data-visualization-with-quantitative-components-77ec53b7ce43]
- 33. Enabling structure-based drug discovery utilizing predicted ... [https://www.sciencedirect.com/science/article/pii/S0092867423014447]
- 34. Next-Generation Sequencing (NGS) | Explore the technology [https://www.illumina.com/science/technology/next-generation-sequencing.html]
- 35. Next-Generation Sequencing Technology: Current Trends ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10376292/]
- 36. Targeted sequencing for plant and animal sciences [https://www.pacb.com/products-and-services/applications/targeted-sequencing/plant-animal-research/]
- 37. Application of Next-Generation Sequencing (NGS ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11171117/]
- 38. Applications of Exome Sequencing in Animals [https://www.cd-genomics.com/resource-exome-sequencing-animals-applications.html]
- 39. An Introduction to Sequence Similarity (“Homology”) Searching [https://pmc.ncbi.nlm.nih.gov/articles/PMC3820096/]
- 40. Alignment-free homology detection | Nature Methods [https://www.nature.com/articles/s41592-024-02470-9]
- 41. 8.2 - Uses of Tabular Data in Bioinformatics | STAT 555 [https://online.stat.psu.edu/stat555/node/84/]
- 42. Protein structure prediction via deep learning: an in-depth ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12003282/]
- 43. Before and after AlphaFold2: An overview of protein ... [https://www.frontiersin.org/journals/bioinformatics/articles/10.3389/fbinf.2023.1120370/full]
- 44. Ten quick tips for homology modeling of high-resolution ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7117658/]
- 45. Advances in protein structure prediction and design [https://www.nature.com/articles/s41580-019-0163-x]
- 46. AlphaFold2 and its applications in the fields of biology and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC10011802/]
- 47. Highly accurate protein structure prediction with AlphaFold [https://www.nature.com/articles/s41586-021-03819-2]
- 48. Advancing protein structure prediction beyond AlphaFold2 [https://www.sciencedirect.com/science/article/abs/pii/S0959440X2500003X]
- 49. Help - SWISS-MODEL [https://swissmodel.expasy.org/docs/help]
- 50. A comparative study of protein structure prediction tools for ... [https://www.sciencedirect.com/science/article/pii/S0041010123003707]
- 51. High-accuracy protein complex structure modeling based ... [https://www.nature.com/articles/s41467-025-65090-7]
- 52. A comparison of profile hidden Markov model procedures for ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC140544/]
- 53. PSI-BLAST Tutorial - Comparative Genomics - NCBI Bookshelf [https://www.ncbi.nlm.nih.gov/books/NBK2590/]
- 54. Deep learning-assisted comparative analysis of animal ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7576204/]
- 55. PSI-BLAST searches using hidden Markov models of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC110734/]
- 56. Automated drug design for druggable target identification ... [https://www.nature.com/articles/s41598-025-18091-x]
- 57. Top-DTI: integrating topological deep learning and large ... [https://pubmed.ncbi.nlm.nih.gov/40662785/]
- 58. Advancing the frontier of rare disease modeling: a critical ... [https://www.nature.com/articles/s41746-025-02068-1]
- 59. Computational nanobody design through deep generative ... [https://www.sciencedirect.com/science/article/pii/S2001037025003125]
- 60. A comparative study of computational modeling ... [https://www.nature.com/articles/s41598-025-16866-w]
- 61. Convergence of Computer-Aided Drug Discovery and ... [https://www.sciencedirect.com/science/article/pii/S2773216925000388]
- 62. Beyond evolution: De novo designed protein toolkit ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12624534/]
- 63. Structure-Based Drug Design: Key Strategies [https://www.saromics.com/structure-based-drug-design-strategies/]
- 64. Structure-based molecular modeling in SAR analysis and lead optimization - PMC [https://pmc.ncbi.nlm.nih.gov/articles/PMC7979990/]
- 65. Computational-aided drug design strategies for drug ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12634332/]
- 66. The Process of Structure-Based Drug Design [https://www.sciencedirect.com/science/article/pii/S1074552103001947]
- 67. Protein Modelling: What Happened to the “Protein Structure ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3816506/]
- 68. Protein Modeling: What Happened to the “Protein Structure ... [https://www.sciencedirect.com/science/article/pii/S0969212613002992]
- 69. Sequence-structure-function relationships in the microbial ... [https://www.nature.com/articles/s41467-023-37896-w]
- 70. Homology Modeling of the mouse MDM2 protein [https://www.bonvinlab.org/education/molmod/modelling/]
- 71. Revisiting gap locations in amino acid sequence ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3110861/]
- 72. A Structured Approach to Optimizing Animal Model Selection ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC9291347/]
- 73. Deep homology of a brachyury cis-regulatory syntax and ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12292651/]
- 74. The dynamic genomes of Hydra and the anciently active ... [https://genomebiology.biomedcentral.com/articles/10.1186/s13059-025-03653-z]
- 75. Translational relevance of forward genetic screens in ... [https://www.sciencedirect.com/science/article/abs/pii/S0149763422000483]
- 76. FDA-NIH Workshop: Reducing Animal Testing - 07/07/2025 [https://www.fda.gov/news-events/fda-meetings-conferences-and-workshops/fda-nih-workshop-reducing-animal-testing-07072025]
- 77. The Impact of Animal Models and Strain Standardization ... [https://www.mdpi.com/2076-393X/13/7/669]
- 78. Empirical statistical estimates for sequence similarity ... [https://www.sciencedirect.com/science/article/abs/pii/S0022283697915254]
- 79. Detecting false positive sequence homology: a machine ... [https://bmcbioinformatics.biomedcentral.com/articles/10.1186/s12859-016-0955-3]
- 80. On the Accuracy of Homology Modeling and Sequence ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC1483079/]
- 81. Harnessing deep learning for proteome-scale detection of ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12261475/]
- 82. Why Is There A Glass Ceiling For Threading Based Protein ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC5398921/]
- 83. Deep homology: a view from systematics. [https://www.biology.ox.ac.uk/publication/53304/pubmed]
- 84. Explainable multi-task learning for multi-modality biological ... [https://www.nature.com/articles/s41467-023-37477-x]
- 85. Multimodal analysis methods in predictive biomedicine [https://pmc.ncbi.nlm.nih.gov/articles/PMC10711035/]
- 86. Fast, sensitive detection of protein homologs using deep ... [https://www.nature.com/articles/s41587-024-02353-6]
- 87. DHR – a rapid pipeline for alignment-free protein homolog ... [https://www.cse.cuhk.edu.hk/news/achievements/prof-yu-lis-group-develops-a-ground-breaking-method-dense-homolog-retrieval-dhr-for-the-rapid-and-sensitive-detection-of-remote-protein-homologs-from-extensive-databases/]
- 88. Improved by 56%, CUHK/Fudan/Yale and... | HyperAI超神经 Sensitivity [https://hyper.ai/en/news/34225]
- 89. xHMMER3x2: Utilizing HMMER 3’s speed and HMMER 2’s sensitivity ... [https://biologydirect.biomedcentral.com/articles/10.1186/s13062-016-0163-0]
- 90. xHMMER3x2: Utilizing HMMER 3's speed and HMMER 2's sensitivity ... [https://pubmed.ncbi.nlm.nih.gov/27894340/]
- 91. Lost in translation: animal models and clinical trials in cancer ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC3902221/]
- 92. One Size Does Not Fit All: The Case for Translational ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6474761/]
- 93. Inflammatory Bowel Disease (IBD)—A Textbook Case for ... [https://www.frontiersin.org/journals/medicine/articles/10.3389/fmed.2019.00230/full]
- 94. Clinical experiences and predictors of success of treatment ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC7821503/]
- 95. Bridging the Gap: Translating Preclinical Biomarkers to ... [https://blog.crownbio.com/bridging-the-gap-translating-preclinical-biomarkers-to-clinical-success]
- 96. Cancer models for reverse and forward translation [https://www.nature.com/articles/s43018-022-00346-5]
- 97. Molecular function limits divergent protein evolution on ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC6750897/]
- 98. Domain-Based and Family-Specific Sequence Identity ... [https://www.sciencedirect.com/science/article/abs/pii/S0022283608015660]
- 99. Local energetic frustration conservation in protein families ... [https://www.nature.com/articles/s41467-023-43801-2]
- 100. New groups of highly divergent proteins in families as old as ... [https://pmc.ncbi.nlm.nih.gov/articles/PMC12153180/]
- 101. Conservation of regulatory elements with highly diverged ... [https://www.nature.com/articles/s41588-025-02202-5]
- 102. Large language models for drug discovery and development [https://pmc.ncbi.nlm.nih.gov/articles/PMC12546459/]
- 103. Principles of early drug discovery - PMC - PubMed Central [https://pmc.ncbi.nlm.nih.gov/articles/PMC3058157/]
- 104. Importance of Target Identification & Validation in Drug ... [https://lifesciences.danaher.com/us/en/library/target-identification-validation-drug-discovery.html]
- 105. Non-Animal & Human-Relevant Research News [https://riseforanimals.org/news/sci-news-sept-2025/]
- 106. Integrative transcriptomic analysis reveals cross - species conserved... [https://bmcpulmmed.biomedcentral.com/articles/10.1186/s12890-025-03928-y]
- 107. Frontiers | Cross - Species of a 6-miRNA Blood Signature... Validation [https://www.frontiersin.org/journals/neurology/articles/10.3389/fneur.2025.1704976/abstract]
- 108. of L-type calcium channel blocker amlodipine as a novel... Validation [https://www.nature.com/articles/s41386-025-02062-x?error=cookies_not_supported&code=73575a23-6144-4e4f-840d-c9b45ba7e8b0]
- 109. Bioinformatic Insights and XGBoost Identify Shared Genetics in... [https://pmc.ncbi.nlm.nih.gov/articles/PMC11882755/]
- 110. Small RNAs and extracellular vesicles: New mechanisms of... [https://journals.plos.org/plospathogens/article?id=10.1371/journal.ppat.1008090]